Beyond the Gene List: Validating Centrality Measures for Powerful ASD Gene Discovery
Autism Spectrum Disorder (ASD) presents immense genetic heterogeneity, challenging the identification of true risk genes.
Beyond the Gene List: Validating Centrality Measures for Powerful ASD Gene Discovery
Abstract
Autism Spectrum Disorder (ASD) presents immense genetic heterogeneity, challenging the identification of true risk genes. This article explores the critical role of network centrality measures in cutting-edge ASD gene discovery pipelines. We first establish the foundational principles of network biology in genomics, then detail methodological applications in machine learning models like forecASD and Stacking-SMOTE. The content addresses key challenges including data imbalance and ancestral diversity bias, offering optimization strategies. Finally, we present a rigorous validation framework, comparing centrality-based predictions against biological evidence from recent studies that define ASD subtypes and their distinct genetic profiles. This synthesis provides researchers and drug developers with a validated, computational roadmap to prioritize novel ASD genes and illuminate underlying biological mechanisms for therapeutic intervention.
The Network Blueprint: Foundational Principles of Centrality in ASD Genetics
Core Concepts: Centrality in Biological Networks
Network theory provides a powerful framework for modeling complex biological systems. In this context, molecules like genes or proteins are represented as nodes, and their physical or functional interactions are represented as edges. Centrality measures are quantitative metrics that assign importance to each node based on its position within the network topology. Their application is crucial for prioritizing key elements, such as candidate disease genes in complex disorders like Autism Spectrum Disorder (ASD) [1] [2] [3].
Frequently Asked Questions
Q: Why would different centrality measures yield different top gene rankings for my ASD dataset? A: Different centrality measures capture distinct topological properties. A systematic survey of 27 centrality measures in protein-protein interaction networks confirmed that the "best" measure depends heavily on the network's specific topology [2]. For instance, Degree centrality identifies highly connected hubs, while Betweenness centrality highlights nodes that connect otherwise separate parts of the network. It is therefore recommended to use a suite of measures and apply Principal Component Analysis (PCA) to identify the most informative ones for your specific biological network [2].
Q: My pathway analysis suggests key genes are "sinks" with no outgoing connections. Why do standard directed centralities rank them as unimportant? A: This is a known limitation of standard directed graph models. In signaling pathways, downstream elements (sinks) are critical receivers of biological signals but may have few or no outgoing edges. The Source/Sink Centrality (SSC) framework addresses this by separately evaluating a node's importance as a sender (Source) and a receiver (Sink) of information, then combining these scores. This method has been shown to more effectively prioritize known cancer and essential genes [3].
Q: How can I validate that my top-ranked centrality genes are biologically relevant to ASD? A: Functional validation is a multi-step process. A common approach is to test for enrichment in known ASD pathways and functions. For example, top genes ranked by game theoretic centrality were enriched for pathways like the immune system, endosomal pathway, and cytokine signaling, all previously implicated in ASD [1]. Furthermore, you can cross-reference your list with high-confidence candidate genes from curated databases like SFARI Gene and check for protein-protein interactions with known ASD genes [1] [4].
Quantitative Comparison of Centrality Measures
The table below summarizes the characteristics of common centrality measures used in biological network analysis, based on a systematic survey in protein-protein interaction networks [2].
Table 1: Key Centrality Measures for Biological Network Analysis
| Centrality Measure | Core Principle | Typical Use Case in Biology | Reported Performance Notes |
|---|---|---|---|
| Degree | Number of direct connections a node has. | Identifying highly connected "hub" proteins; correlates with essentiality [2]. | Simple but effective; performance can be variable across networks [2]. |
| Betweenness | Number of shortest paths that pass through a node. | Finding bottleneck proteins that connect functional modules [2]. | Often outperforms Degree in modular networks [2]. |
| Closeness | Average shortest path distance from a node to all others. | Identifying nodes that can quickly influence the entire network. | High contribution across diverse networks; several variants exist [2]. |
| PageRank | Measures node influence based on the influence of its neighbors. | Ranking genes in pathways; a random walk with restart model [3]. | Standard directed version undervalues sink nodes [3]. |
| Subgraph | Measures node importance based on its participation in all subgraphs. | Identifying structurally central proteins. | Outperformed classic measures in early essentiality studies [2]. |
| Game Theoretic (Shapley Value) | Evaluates a node's marginal contribution to all possible coalitions. | Prioritizing genes based on synergistic influence in a network [1]. | Novel approach that can highlight genes missed by other measures [1]. |
Experimental Protocols
Protocol: Implementing a Game Theoretic Centrality Analysis for ASD Gene Discovery
This protocol is adapted from studies that used the Shapley value to prioritize disease genes by combining biological networks with coalitional game theory [1].
1. Objective: To rank genes by their synergistic influence in a gene-to-gene interaction network and prioritize candidate genes for ASD.
2. Research Reagent Solutions
Table 2: Essential Materials for Game Theoretic Centrality Analysis
| Item | Function / Explanation | Example Source |
|---|---|---|
| Biological Network | Provides the graph structure for analysis. Represents gene-gene interactions. | STRING database (protein-protein interactions) [1]. |
| Genetic Dataset | The set of genes to be analyzed and ranked. | Whole genome sequence data from multiplex autism families [1]. |
| Gold-Standard ASD Genes | A set of high-confidence genes for validation and model benchmarking. | SFARI Gene database [1]. |
| Pathway Analysis Tool | To biologically validate top-ranking genes by testing for enrichment in known processes. | Reactome Pathway Browser [1]. |
3. Step-by-Step Workflow:
- Step 1 - Network Construction: Obtain a protein-protein interaction (PPI) network from a database like STRING. This network defines the set of players (genes) for the coalitional game.
- Step 2 - Define the Characteristic Function: For any coalition (subset) of genes
S, the characteristic functionv(S)quantifies the coalition's "worth." This is often defined based on the network's connectivity, for example, the number of nodes outsideSthat are connected to nodes within it. - Step 3 - Calculate Shapley Value: For each gene
i, compute its Shapley value. The Shapley value is the weighted average of the gene's marginal contributionv(S ∪ {i}) - v(S)across all possible coalitionsS. This calculation is computationally intensive and often requires approximation algorithms for large networks. - Step 4 - Rank Genes: Rank all genes in descending order of their Shapley value. Genes with the highest scores are those that, on average, contribute the most to the connectivity of their neighbors.
- Step 5 - Biological Validation:
- Cross-reference: Compare top-ranked genes with known ASD gene sets (e.g., from SFARI Gene).
- Pathway Enrichment: Use a tool like the Reactome Pathway Browser to test if top-ranked genes are significantly enriched for biological pathways previously linked to ASD (e.g., immune system, synaptic signaling) [1].
- Literature Mining: Manually check the association of top novel candidates with ASD or other neurodevelopmental disorders in the literature.
Protocol: Building a Tissue-Specific Network for Omnigenic Analysis
This protocol is based on research that used tissue-specific networks to study the omnigenic model in ASD, which distinguishes core genes from peripheral genes [4].
1. Objective: To construct and analyze a tissue-specific gene interaction network to identify core and peripheral gene clusters relevant to ASD.
2. Research Reagent Solutions
- Tissue-Specific Networks: Genome-scale Integrated Analysis of gene Networks in Tissues (GIANT) database [4].
- Core Gene Set: A curated list of putative core ASD genes (e.g., SFARI Gene database, excluding the lowest confidence scores) [4].
- Clustering Algorithm: Louvain method for community detection [4].
3. Step-by-Step Workflow:
- Step 1 - Network Selection: Download a tissue-specific gene interaction network from the GIANT database. For ASD, the most relevant network is typically derived from brain tissue.
- Step 2 - Extract Core Subgraph: Map your curated set of core ASD genes (e.g., from SFARI) onto the full network. Extract the subgraph that includes only these core genes and the edges between them.
- Step 3 - Calculate Node Strength: For each core gene, calculate its node strength in the full tissue-specific network. Node strength is the sum of the weights of all edges connected to a node, indicating its overall connectedness.
- Step 4 - Cluster Analysis: Apply the Louvain clustering algorithm to the full network to identify communities (clusters) of tightly connected genes.
- Step 5 - Interpret Clusters: Biologically interpret the resulting clusters by performing Gene Ontology enrichment analysis on the genes within each cluster. Clusters in brain tissue are expected to be enriched for functions like synaptic signaling and chromatin remodeling [4].
Visualizations
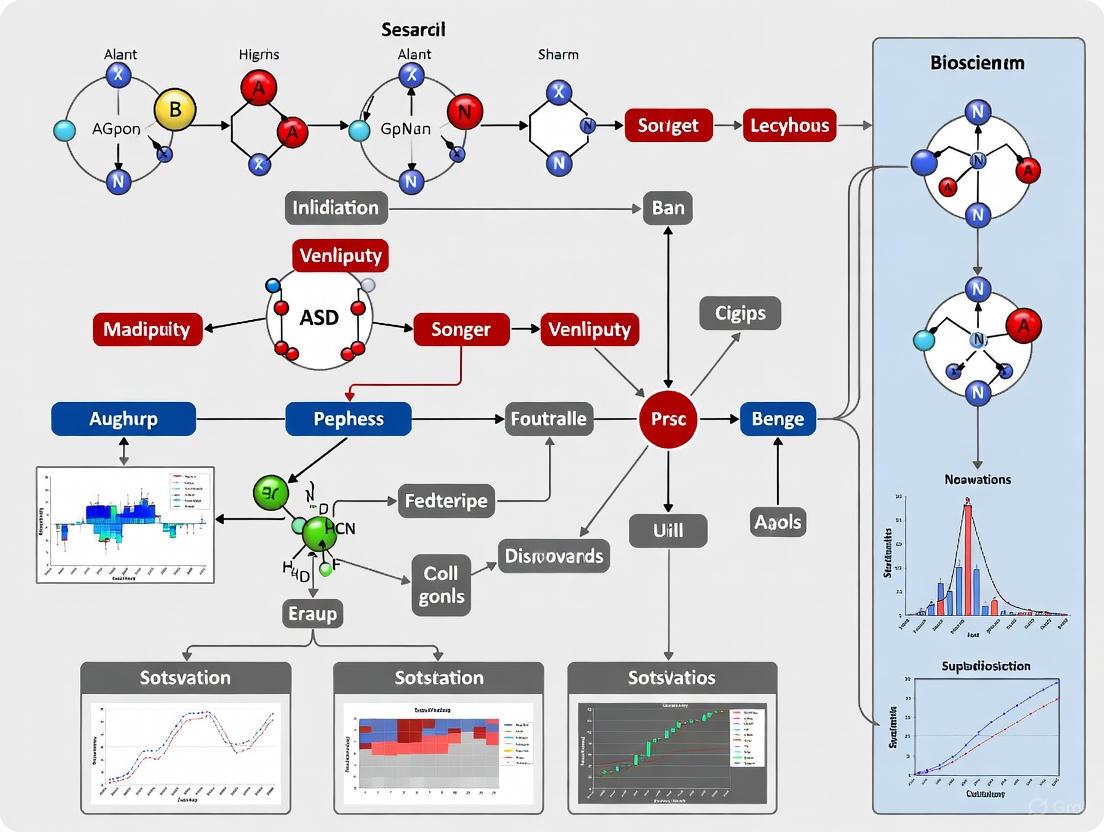
Centrality Analysis Workflow
The diagram below illustrates the integrated workflow for identifying and validating ASD risk genes using network centrality measures, as described in the experimental protocols.
Source/Sink Centrality Framework
This diagram contrasts standard directed centrality with the Source/Sink Centrality (SSC) framework, which is critical for accurately modeling biological pathways.
Omnigenic Model in Tissue Networks
This visualization depicts the core-periphery structure of the omnigenic model within a tissue-specific gene interaction network.
Troubleshooting Common Research Challenges
FAQ: Why do single-gene approaches have limited success in ASD research? ASD is characterized by extreme genetic heterogeneity, with hundreds of genes implicated and most individual genes accounting for less than 0.5% of cases [5] [6]. The genetic architecture involves both rare variants with strong effects and common variants with weak effects working in combination [7] [6]. Single-gene approaches cannot capture this polygenic complexity or the gene-gene interactions that contribute to ASD pathophysiology.
FAQ: How can researchers account for the clinical heterogeneity in ASD genetic studies? Recent studies have adopted data-driven subtyping approaches that integrate phenotypic and genotypic data. One 2025 study analyzing over 5,000 ASD individuals identified four distinct classes with different biological signatures: Social and Behavioral Challenges (37%), Mixed ASD with Developmental Delay (19%), Moderate Challenges (34%), and Broadly Affected (10%) [8]. These subtypes show minimal overlap in impacted biological pathways, suggesting different underlying mechanisms [8].
FAQ: What biological pathways are consistently implicated across ASD genetic studies? Despite genetic heterogeneity, ASD risk genes converge on several key biological processes as shown in the table below:
Table: Key Biological Pathways Implicated in ASD
| Pathway Category | Specific Pathways | Representative Genes |
|---|---|---|
| Synaptic Function | Synaptic formation, neurotransmitter signaling, neural connectivity | NLGN3, NLGN4X, NRXN1, SHANK3 [5] [7] |
| Chromatin & Transcription | Chromatin remodeling, transcriptional regulation, epigenetic modification | CHD8, MECP2, ADNP, FMRP [5] [7] |
| Immune System | Immune system, cytokine signaling, HLA complex | HLA-A, HLA-B, HLA-G, HLA-DRB1 [1] |
FAQ: How do genetic modifiers influence ASD presentation? Genetic modifiers including copy number variations, single nucleotide polymorphisms, and epigenetic alterations can significantly modulate the phenotypic spectrum of ASD patients with similar pathogenic variants [7]. For example, individuals with similar 15q duplications can present from unaffected to severely disabled [7]. These modifiers likely alter convergent signaling pathways and lead to impaired neural circuitry formation through complex interactions [7].
Experimental Protocols for Advanced ASD Gene Discovery
Hybrid Deep Learning for Key Gene Identification
Table: Research Reagent Solutions for Network Analysis
| Research Reagent | Function/Application | Source |
|---|---|---|
| Autism Informatics Portal ASD Gene Set | Provides comprehensive list of ASD-associated genes for network construction | [9] |
| STRING Database | Constructs protein-protein interaction networks restricted to Homo sapiens | [9] |
| Graph Convolutional Network (GCN) | Extracts node embeddings from PPI networks based on topological features | [9] |
| Centrality Measures (DC, BC, CC, EC) | Quantifies node importance in biological networks for feature matrix | [9] |
Protocol: Hybrid Deep Learning Approach to Identify Key ASD Genes
Sample Preparation & Data Collection:
- Data Acquisition: Obtain the comprehensive list of ASD genes (n=1,215) from the Autism Informatics Portal [9].
- Data Curation: Remove duplicates, isolated, and redundant nodes to yield a final dataset (recommended: 979 genes) [9].
- Network Construction: Use STRING database to construct a protein-protein interaction network restricted to Homo sapiens, typically resulting in ~9,505 interactions among the ASD genes [9].
Feature Processing:
- Network Representation: Create an undirected graph G=(V,E,A) where V represents nodes (genes), E represents edges (interactions), and A is the adjacency matrix [9].
- Feature Matrix Calculation: Compute the feature matrix X based on various topological properties:
- Degree centrality: Measures direct connections [9]
- Betweenness centrality: Identifies nodes on shortest paths [9]
- Closeness centrality: Quantifies information spread efficiency [9]
- Eigenvector centrality: Measures influence based on neighbors' importance [9]
- Clustering coefficient: Assesses interconnectivity of neighbors [9]
Model Implementation:
- Input Layer: Feed the ASD network (nodes and links) into the model [9].
- Feature Processing Layer: Extract neighbors for each node and form adjacency matrix [9].
- GCN Architecture: Apply graph convolutional networks to extract node embeddings [9].
- Logistic Regression Layer: Predict potential key regulator genes using probability scores (0-1 range) [9].
Validation:
- Evaluation: Use susceptible-infected (SI) model to evaluate infection ability of potential key regulator genes [9].
- Comparison: Validate against established databases like SFARI Gene and EAGLE framework [9].
Game Theoretic Centrality for Gene Prioritization
Protocol: Coalitional Game Theory Approach for ASD Gene Ranking
Data Preparation:
- Cohort Selection: Utilize multiplex autism families (recommended: 756 families with 1,965 children) [1].
- Variant Identification: Identify likely gene-disrupting (LGD) variants from whole genome sequence data [1].
Network Integration:
- Biological Network Construction: Incorporate a priori knowledge from biological networks including protein-protein interaction data and pathway information [1].
- Graph Formation: Create computable networks representing various biological systems [1].
Game Theoretic Analysis:
- Coalition Formation: Evaluate coalitions that form among genes and find players that marginally contribute the most on average [1].
- Shapley Value Calculation: Apply game theoretic centrality measure based on Shapley value to rank genes by their relevance in the gene-to-gene interaction network [1].
- Topological Assessment: Explore topological properties of biological networks to study combinatorial effects [1].
Validation & Pathway Analysis:
- Biological Validation: Cross-reference top ranking genes with established ASD gene databases (SFARI, Root 66 gene list) [1].
- Pathway Enrichment: Use Reactome Pathway Browser to identify significant pathways enriched in top ranking genes [1].
- Protein Interaction Checking: Identify direct protein-protein interactions between game theoretic centrality genes and high-confidence candidate ASD genes [1].
Quantitative Data Synthesis
Table: Detection Rates of Genetic Abnormalities in ASD Populations
| Genetic Testing Approach | Detection Rate in ASD | Key Findings | Clinical Utility |
|---|---|---|---|
| Chromosomal Microarray (CMA) | ~7-10% [5] | Identifies rare or de novo CNVs; reveals recurrent CNV hotspots (1q21.1, 15q13.3, 16p11.2) [5] | First-tier test for non-specific ASD [5] |
| Whole Exome Sequencing | Varies by study [6] | Hundreds of candidate genes identified; most account for <0.5% of cases individually [6] | Identifies de novo mutations in sporadic cases [6] |
| Hybrid Deep Learning | Superior to centrality methods [9] | Higher infection ability for identified genes; aligns with SFARI database [9] | Pinpoints key genetic factors from complex networks [9] |
Table: ASD Subtypes with Distinct Genetic Profiles Identified in Recent Studies
| ASD Subtype | Prevalence | Developmental Trajectory | Genetic Correlations | Key Biological Pathways |
|---|---|---|---|---|
| Social & Behavioral Challenges | 37% [8] | Few developmental delays; later diagnosis [8] | Moderate correlation with ADHD/mental health conditions [10] | Postnatally active genes; neuronal function [8] |
| Mixed ASD with Developmental Delay | 19% [8] | Early developmental delays [8] | Lower correlation with ADHD/mental health conditions [10] | Prenatally active genes; chromatin organization [8] |
| Moderate Challenges | 34% [8] | Variable presentation [8] | Intermediate genetic profile [8] | Mixed pathway involvement [8] |
| Broadly Affected | 10% [8] | Widespread challenges across domains [8] | Complex polygenic architecture [8] | Multiple disrupted pathways [8] |
Frequently Asked Questions
1. What is network centrality and why is it important in biological research? Network centrality is a fundamental concept in network analysis that measures the importance or influence of a node (e.g., a gene or protein) within a network. Importance is defined in different ways, leading to different centrality measures [11]. In biological research, such as ASD gene discovery, centrality helps identify essential nodes. These often correspond to genes that are more likely to be associated with indispensability or disease risk when disrupted [12]. Analyzing centrality allows researchers to move beyond simple gene lists to understanding genes' roles within the complex web of molecular interactions [13] [1].
2. How do I choose the right centrality measure for my gene network analysis? The choice depends on the specific biological question you are investigating. The table below summarizes the core applications of three key measures in a biological context:
| Centrality Measure | Best Used For |
|---|---|
| Degree Centrality | Identifying genes with many direct interactions (hubs), which are often critical for network stability and can be essential for survival [12] [14]. |
| Betweenness Centrality | Finding bottleneck genes that control information or flow between different network modules. These are potential key regulators in signaling pathways [12] [15]. |
| Eigenvector Centrality | Pinpointing influential genes that are connected to other highly influential genes, suggesting they are part of a central, tightly-knit core complex or pathway [16] [11]. |
3. A known ASD risk gene has a low centrality score in my analysis. Does this mean it's unimportant? Not necessarily. The network's structure and the specific measure used affect results [11]. A gene might have low degree but be functionally critical. It is recommended to use multiple centrality measures and integrate other biological evidence (e.g., gene expression, functional annotations) to get a comprehensive view [12]. Some methods, like game-theoretic centrality, are specifically designed to identify genes that are influential within their local neighborhood rather than the entire network, which may capture important but less globally central genes [1].
4. My betweenness centrality calculations are computationally expensive. Are there efficient alternatives? Yes, computational cost can be a challenge for large networks. While betweenness centrality relies on calculating all shortest paths [16], other measures can provide valuable insights more efficiently. Degree centrality is the fastest to compute [11]. Alternatively, consider using closeness centrality, which identifies nodes that can efficiently reach all other nodes by calculating the inverse of the sum of the shortest paths to all other nodes [16]. For very large networks, investigate approximate algorithms for betweenness calculation or leverage game-theoretic centrality, which has been successfully applied to large genomic datasets [1].
Experimental Protocols & Methodologies
Protocol 1: Calculating Centrality Measures for a Protein-Prointeraction Network (PPI)
This protocol outlines the steps to calculate and interpret centrality measures from a PPI network to prioritize candidate genes.
- Network Acquisition and Construction: Obtain a high-quality, context-specific PPI network. Sources like STRING or BioGRID are common starting points. For ASD research, prioritize networks derived from brain tissue or neurodevelopmental stages [13] [1].
- Data Preprocessing: Filter the network to remove low-confidence interactions and ensure the largest connected component is used for analysis to allow calculation of path-based measures like betweenness and closeness.
- Centrality Calculation: Use network analysis tools (e.g., igraph in R, NetworkX in Python) to compute the three centrality measures for every node.
- Gene Ranking and Integration: Rank genes based on each centrality score. Compare these rankings with known ASD gene sets (e.g., from SFARI database) for validation [1]. Finally, integrate the results with other genomic data, such as gene expression from the BrainSpan atlas or gene-level constraint metrics (e.g., pLI scores), to strengthen predictions [13] [12].
The workflow for this protocol is summarized in the following diagram:
Protocol 2: Integrating Centrality with Machine Learning for Gene Prediction
This advanced protocol leverages centrality as a feature in a machine learning model to predict novel ASD risk genes, as demonstrated in contemporary studies [13].
- Define Training Set: Curate a set of known positive genes (e.g., high-confidence ASD genes from SFARI) and true negative genes (genes associated with non-neurological diseases) [13].
- Feature Extraction: For each gene, calculate multiple features. This includes:
- Topological Features: Degree, betweenness, and eigenvector centrality from a biological network [13].
- Gene Constraint Metrics: pLI, LOEUF scores from sources like gnomAD, which quantify a gene's intolerance to mutations [13].
- Spatiotemporal Expression: Features derived from brain gene expression data across development (e.g., from BrainSpan atlas) [13].
- Model Training and Prediction: Train a supervised machine learning classifier (e.g., Random Forest) on the labeled gene set. Use the trained model to score and rank novel genes.
The logical flow of this machine learning approach is shown below:
Quantitative Data on Centrality in Biology
The table below synthesizes key findings from research on the application of centrality measures in biological networks, highlighting their utility and limitations.
| Centrality Measure | Correlation with Essentiality | Key Findings and Biological Interpretation |
|---|---|---|
| Degree Centrality | Variable, often positive | Correlates with lethality in some organisms (e.g., yeast) but not always (e.g., E. coli metabolic networks). High-degree nodes are "hubs" whose disruption can destabilize the network [12]. |
| Betweenness Centrality | Positive in many studies | Identifies "bottleneck" nodes. In drug networks, high-betweenness drugs are better candidates for triggering drug repositioning [15]. In PPI networks, it correlates with essentiality [12]. |
| Eigenvector Centrality | Positive | Highlights nodes connected to other influential nodes. It is part of a family of measures that consider a node's connection to important neighbors, making it effective at finding central nodes in a connected core [11] [12]. |
| Combined Measures | Improved Performance | Combining centralities (e.g., degree and closeness) can yield more reliable predictions of essential genes than any single measure [12]. Game-theoretic centrality also identifies influential genes missed by standard measures [1]. |
The Scientist's Toolkit: Research Reagent Solutions
| Item / Resource | Function in Centrality Analysis |
|---|---|
| STRING Database | A database of known and predicted Protein-Protein Interactions (PPIs) used to construct the underlying network for analysis [1]. |
| igraph / NetworkX | Open-source software libraries (in R and Python, respectively) used to calculate centrality measures and perform network analysis [16]. |
| BrainSpan Atlas | A resource of spatiotemporal human brain gene expression data. Used to create co-expression networks or validate that candidate genes are active in relevant brain regions and developmental windows [13]. |
| ExAC/gnomAD | Databases providing gene-level constraint metrics (e.g., pLI scores). These quantify a gene's intolerance to loss-of-function mutations and serve as valuable features to integrate with topological data [13]. |
| SFARI Gene Database | A curated resource of genes associated with Autism Spectrum Disorder. Used as a benchmark "gold standard" set for validating and prioritizing genes identified through centrality analysis [13] [1]. |
Frequently Asked Questions (FAQs)
1. What does a high "degree centrality" score indicate about a gene or protein? A high degree centrality indicates that a gene or protein is a hub in the network, meaning it has a large number of direct interactions with other molecules [17] [18]. Biologically, this often suggests the molecule plays a fundamental, housekeeping role and is involved in key regulatory functions or serves as a critical connector in cellular processes. In protein interaction networks, such hubs are often essential for survival, and their disruption can be lethal [17].
2. How is "betweenness centrality" biologically interpreted? A high betweenness centrality score identifies nodes that act as critical bottlenecks in the network [17]. These genes or proteins often reside on many of the shortest paths between other pairs of nodes, meaning they control the information flow or communication between different network modules. This can indicate a role in coordinating signals between otherwise separate biological processes. Proteins with high betweenness but low connectivity (HBLC proteins) are particularly interesting as they may support network modularization [17].
3. My analysis shows a gene has high "closeness centrality." What does this mean? A gene with high closeness centrality can, on average, reach all other genes in the network in a relatively small number of steps [17]. This suggests it is a highly influential node, positioned to rapidly affect the state of the entire network or to quickly gather information from across the network. In metabolic networks, for example, metabolites with high closeness are often part of central pathways like glycolysis and the citrate acid cycle [17].
4. Why should I use multiple centrality metrics in my analysis? Different centrality metrics highlight nodes with different functional roles [19]. Relying on a single metric provides a limited view, as a node can be central in one aspect (e.g., a local hub with high degree) but not in another (e.g., a global bottleneck with high betweenness). Using multiple metrics—such as degree, betweenness, and closeness—offers a more comprehensive and accurate assessment of a node's importance from various structural perspectives [20] [19].
5. How are centrality measures applied in the context of Autism Spectrum Disorder (ASD) research? In ASD research, centrality-based pathway enrichment methods help identify significant biological pathways dominated by key genes [20]. This is crucial for parsing the extreme genetic and phenotypic heterogeneity of ASD. By applying centrality analysis to gene networks, researchers can pinpoint biologically meaningful subtypes of ASD, linking distinct phenotypic classes (e.g., "Social/Behavioral Challenges," "Mixed ASD with Developmental Delay") to specific underlying genetic programs and disrupted biological pathways [21] [22].
Troubleshooting Guides
Issue 1: Interpreting Contradictory Centrality Scores
Problem: A gene has a high score for one centrality measure (e.g., high degree) but a low score for another (e.g., low betweenness). It is unclear how to interpret its biological importance.
Solution:
- Interpret the Functional Role: This pattern is common and reveals a specific network role. A high-degree, low-betweenness node is typically a local hub within a dedicated functional module, but it does not control flow between modules. Conversely, a low-degree, high-betweenness node acts as a critical bridge or bottleneck connecting different parts of the network [17] [19].
- Cross-Reference with Biological Knowledge: Integrate the structural findings with existing annotation. Check if the gene is known to be part of a large protein complex (consistent with high degree) or a key signaling intermediary (consistent with high betweenness).
- Consult a Decision Matrix: Use the following table to guide your interpretation.
| Centrality Profile | Structural Role | Proposed Biological Interpretation | Common in ASD-Related Pathways? |
|---|---|---|---|
| High Degree, Low Betweenness | Local hub within a module | Core component of a stable complex or a central enzyme in a metabolic pathway. Essential for a specific, localized function. | Yes, e.g., genes within synaptic scaffolding complexes. |
| Low Degree, High Betweenness | Global bottleneck, bridge | Key regulatory molecule, signaling intermediary, or transcription factor that integrates information from multiple pathways. | Yes, e.g., high-betweenness genes connecting neurodevelopmental pathways [17]. |
| High Closeness | Centrally located influencer | A molecule with broad, rapid influence over the network state, potentially a master regulator. | Seen in genes regulating early brain development. |
| High Degree, High Betweenness | Central hub and bottleneck | A molecule of critical, multi-faceted importance. Its disruption is highly likely to have severe, system-wide consequences. | Often found among high-confidence ASD risk genes. |
Issue 2: Integrating Centrality Analysis with Differential Expression for ASD Gene Discovery
Problem: A list of differentially expressed genes (DEGs) from an ASD case-control study has been generated, but it is challenging to prioritize them for functional validation.
Solution: Implement a Centrality-Based Pathway Enrichment Workflow. This method moves beyond simple gene counting by incorporating the topological structure of biological pathways [20].
Experimental Protocol: Centrality-Based Pathway Analysis
Objective: To identify pathways not just enriched with DEGs, but dominated by topologically central DEGs, which may have greater functional impact.
Methodology:
- Input Data:
- Gene Expression Matrix: From your RNA-seq or microarray experiment (e.g., ASD vs. control).
- Pathway Database: A curated source with topological information, such as the NCI-Nature PID, KEGG, or Reactome [20].
- Map Genes to Pathway Nodes:
- For each pathway, convert the gene set into a graph
G = (V, E), whereVis a set of nodes (proteins, complexes) andEis a set of edges (interactions, reactions) [17] [20]. - Map your DEGs onto these pathway nodes. If any gene in a multi-gene complex node is differentially expressed, mark that node as "affected" [20].
- For each pathway, convert the gene set into a graph
- Calculate Node Centrality:
- Compute a Weighted Pathway Score:
- Instead of a simple count, calculate a pathway significance score (e.g., a modified Fisher's exact test or a Mann-Whitney U test) that weights each DEG by its centrality score within the pathway. This gives more importance to differentially expressed genes that are central hubs or bottlenecks [20].
- Statistical Significance:
- Determine significance of the weighted pathway score through permutation testing, creating a null distribution by randomly shuffling gene labels.
Workflow for Centrality-Based Pathway Analysis
Issue 3: Validating Centrality Findings in a Neurodevelopmental Context
Problem: After identifying high-centrality genes, you need a biologically relevant way to validate their functional importance in neurodevelopment, particularly for ASD.
Solution: Leverage person-centered phenotypic subclassification and single-cell transcriptomic data. Recent large-scale studies provide a framework for linking network topology to clinical and molecular data [21] [22].
Experimental Protocol: Functional Validation of High-Centrality ASD Genes
Objective: To test whether high-centrality genes from your analysis are enriched in specific ASD subtypes and expressed in relevant neuronal cell types.
Methodology:
- Subtype Association Analysis:
- Obtain the list of high-centrality genes from your network analysis.
- Using a dataset like SPARK (n=5,392 individuals), test for enrichment of damaging mutations (e.g., de novo protein-truncating variants) in your gene set within the defined phenotypic subtypes (e.g., "Broadly Affected," "Social/Behavioral") [21] [22].
- Statistical Test: Use a gene-based burden test (e.g., TADA) comparing the rate of mutations in your gene set between subtypes and controls [23] [21].
- Temporal Expression Analysis:
- Use developmental transcriptome data (e.g., BrainSpan Atlas of the Developing Human Brain) to analyze the expression trajectory of your high-centrality genes.
- Test if they are enriched in co-expression modules specific to particular developmental periods (e.g., mid-fetal prefrontal cortex) [21].
- Single-Cell Resolution:
Functional Validation Strategy for ASD Genes
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function/Biological Interpretation |
|---|---|
| R Statistical Environment & CePa Package [20] | A software platform and specific package for performing centrality-based pathway enrichment analysis, allowing for the integration of topological information into gene set testing. |
| Pathway Interaction Database (PID) [20] | A curated database of biomolecular interactions and pathways, often used for centrality analysis because it includes information on protein complexes and signaling networks. |
| Protein-Protein Interaction (PPI) Data (e.g., from STRING, BioGRID) [18] | Raw data used to construct the networks on which centrality is calculated. Represents physical or functional associations between proteins. |
| Gene Set Enrichment Analysis (GSEA) Software [20] | A foundational tool for gene set analysis. Centrality-based methods can be viewed as an extension that adds node-weighting to the GSEA procedure. |
| Transmission and De Novo Association (TADA) Model [23] [21] | A Bayesian statistical framework that integrates de novo and rare inherited variants to identify genes with a significant burden of mutations in disease cohorts like ASD. Used for gene discovery and validation. |
| BrainSpan Atlas Data | A resource of developmental transcriptome data from post-mortem human brains, used to validate the temporal expression patterns of high-centrality genes. |
| Single-Cell RNA-Seq Datasets (e.g., from developing human cortex) [23] [21] | Data used to confirm that high-centrality genes are expressed in specific, disease-relevant neuronal cell types at critical developmental time points. |
Troubleshooting Guides
Guide 1: Addressing Low Specificity in Your PPI Network
Problem: The constructed Protein-Protein Interaction (PPI) network is too large and non-specific, containing a high fraction of human genes, which dilutes potential ASD-relevant signals [24].
Solution:
- Step 1: Filter interactions based on brain-specific expression data. Incorporate gene expression data from resources like the Human Protein Atlas (HBTB RNA-seq data) to focus on genes expressed in relevant brain tissues [24].
- Step 2: Validate network specificity using a Monte-Carlo approach. Randomly sample genes from the HGNC database (using 1000 random seeds) and compare SFARI gene enrichment in your network against random networks. A statistically significant enrichment (e.g., p < 2.2×10⁻¹⁶) confirms network specificity [25] [24].
- Step 3: Apply spatiotemporal expression filters from brain development datasets to ensure biological relevance to neurodevelopment [24].
Guide 2: Overcoming Hub Gene Bias in Centrality Measures
Problem: Betweenness centrality and other centrality measures tend to highlight highly connected hub genes that may not be specifically relevant to ASD pathophysiology [24].
Solution:
- Step 1: Combine multiple centrality measures. Implement game theoretic centrality (Shapley value) which considers synergistic gene influences and may identify different candidates compared to traditional measures [26] [1].
- Step 2: Integrate functional validation data. Correlate centrality rankings with:
- De novo mutation data from ASD cohorts
- RNA-seq data from ASD brain tissue
- Co-expression patterns with known ASD genes [24]
- Step 3: Perform pathway enrichment analysis on top-ranked genes to verify biological relevance to known ASD mechanisms [25] [1].
Guide 3: Validating Centrality-Based Gene Prioritization
Problem: How to determine if centrality-prioritized genes are genuinely relevant to ASD rather than statistical artifacts.
Solution:
- Step 1: Cross-reference with established ASD gene databases including SFARI Gene, "Root 66" differentially expressed genes, and rare variant genes [26] [1].
- Step 2: Conduct protein-protein interaction checks between prioritized genes and high-confidence ASD genes using STRING database [1].
- Step 3: Perform functional enrichment analysis using Reactome Pathway Browser to identify if prioritized genes converge on pathways previously implicated in ASD (e.g., immune system, synaptic function) [1].
Frequently Asked Questions (FAQs)
FAQ 1: Which centrality measure performs best for ASD gene discovery? Answer*: Current evidence suggests that different centrality measures identify complementary gene sets:
- Betweenness centrality: Effective for identifying bottleneck proteins in PPI networks; correlated with other topological metrics [25]
- Game theoretic centrality: Identifies influential genes that might be missed by traditional measures; only 10-20% overlap with betweenness centrality rankings [26] [1]
- Graph neural networks: Graph Sage models achieve 85.80% accuracy in binary risk classification [27]
Table: Comparison of Centrality Measures for ASD Gene Prioritization
| Centrality Measure | Key Principle | Performance/Advantages | Limitations |
|---|---|---|---|
| Betweenness Centrality | Identifies nodes that frequently lie on shortest paths between other nodes | Correlated with other topological metrics; effectively prioritizes genes in noisy datasets [25] | Tendency to highlight general hub genes not specific to ASD [24] |
| Game Theoretic Centrality | Based on Shapley value; evaluates marginal contribution of genes in networks | Identifies distinct genes (e.g., HLA complex, ATP6AP1); reveals immune pathways in ASD [26] [1] | Limited to well-annotated protein-coding genes; misses non-coding regions [1] |
| Graph Neural Networks (Graph Sage) | Uses machine learning on gene networks with chromosome location features | 85.80% accuracy for binary risk classification; 81.68% for multi-class risk [27] | Requires substantial computational resources and training data [27] |
FAQ 2: How can I improve my PPI network's relevance to ASD? Answer*: Implement a multi-step filtering approach:
- Start with high-confidence ASD genes from SFARI (scores 1-2) as seed nodes [25]
- Extract first interactors from IMEx database to build initial network [25]
- Filter for brain-expressed genes using Human Protein Atlas data (94.3% of nodes in validated networks maintain brain expression) [24]
- Validate enrichment significance against randomly generated networks [25]
FAQ 3: What are the most common pitfalls when applying centrality measures to ASD networks? Answer*: The main pitfalls include:
- Overly large networks: Including ~63% of human protein-coding genes reduces specificity [24]
- Hub gene bias: Highly connected genes may be biologically essential but not ASD-specific [24]
- Ignoring syndromic genes: Separate analysis of syndromic vs. non-syndromic genes improves accuracy (Graph Sage models achieve 90.22% accuracy on syndromic classification) [27]
- Lacking experimental validation: Always complement computational findings with expression data, mutation evidence, or functional studies [24] [1]
Experimental Protocols
Protocol 1: Building a Prioritized ASD Gene Network Using Betweenness Centrality
Purpose: To construct and analyze a protein-protein interaction network for prioritizing ASD-associated genes using betweenness centrality [25].
Materials:
- SFARI Gene database (access via https://gene.sfari.org/) [28]
- IMEx database for protein-protein interactions [25]
- Human Protein Atlas for brain expression data [24]
- Network analysis software (e.g., Cytoscape, NetworkX)
Procedure:
- Data Collection:
Network Construction:
Topological Analysis:
- Calculate betweenness centrality for all nodes using formula:
- BC(v) = Σs≠v≠t σst(v)/σst
- Where σst is total shortest paths from node s to t, and σst(v) is number of those passing through v [25]
- Rank genes by decreasing betweenness centrality score
- Calculate betweenness centrality for all nodes using formula:
Validation:
- Perform Monte-Carlo simulation: randomly select 12,598 protein-coding genes from HGNC database 1000 times
- Compare SFARI gene enrichment in your network vs. random networks using one-sample t-test
- Statistically significant enrichment: p-value < 2.2×10⁻¹⁶ [25]
Troubleshooting:
- If network is too large (>13,000 nodes), apply additional brain-specific expression filters
- If betweenness centrality identifies mostly general hub genes, integrate with spatiotemporal expression data [24]
Protocol 2: Implementing Game Theoretic Centrality for ASD Gene Discovery
Purpose: To apply game theoretic centrality based on Shapley value to prioritize influential ASD genes within biological networks [26] [1].
Materials:
- Whole genome sequence data from multiplex autism families [26]
- STRING database for protein-protein interactions [1]
- Coalitional game theory implementation code [26]
Procedure:
- Network Preparation:
- Build a PPI network using data from STRING database
- Include both well-annotated protein-coding genes and any available pseudogene data [26]
Game Theoretic Analysis:
Biological Validation:
- Cross-reference top-ranking genes with:
- SFARI Gene database
- "Root 66" differentially expressed gene set
- Rare variant genes from recent ASD studies [1]
- Perform pathway enrichment analysis using Reactome Pathway Browser
- Focus on pathways with FDR < 0.05, particularly immune system and neuronal function pathways [1]
- Cross-reference top-ranking genes with:
Expected Results:
- Top-ranked genes should include HLA complex genes (HLA-A, HLA-B, HLA-G, HLA-DRB1) [1]
- Enrichment in immune system pathways (FDR = 2.15×10⁻¹⁵) and endosomal pathways (FDR = 2.15×10⁻¹⁵) [1]
- Approximately 10-20% overlap with betweenness centrality results [26]
Signaling Pathways and Experimental Workflows
Centrality Integration Workflow for ASD Gene Discovery
Diagram Title: Centrality Integration Workflow for ASD Gene Discovery
ASD Gene Network Centrality Pathways
Diagram Title: ASD Gene Network Centrality Pathways
Research Reagent Solutions
Table: Essential Research Reagents and Databases for Centrality-Based ASD Research
| Research Reagent/Database | Type | Primary Function in ASD Network Analysis | Key Features/Applications |
|---|---|---|---|
| SFARI Gene Database | Curated database | Provides validated ASD-associated genes for network seeding | Contains gene scores (1-3 confidence levels); syndromic/non-syndromic classification; regularly updated [28] |
| IMEx Database | Protein-protein interaction database | Source of experimentally validated physical interactions for PPI network construction | International consortium data; curated physical interactions; includes multiple organism data [25] |
| Human Protein Atlas | Tissue expression database | Filtering network nodes based on brain-specific expression | RNA-seq data from brain tissues; allows specificity refinement of PPI networks [24] |
| STRING Database | PPI database | Alternative source for protein interaction data | Includes both experimental and predicted interactions; useful for game theoretic centrality [1] |
| Reactome Pathway Browser | Pathway analysis tool | Functional enrichment analysis of prioritized genes | Identifies significantly enriched pathways; FDR correction; connects genes to biological processes [1] |
| ABIDE Dataset | Neuroimaging database | Validation of network findings against brain connectivity data | Resting-state fMRI data from ASD and control subjects; correlation with structural findings [29] |
From Theory to Tool: Methodological Integration of Centrality in Discovery Pipelines
Frequently Asked Questions (FAQs)
Q1: What is the core premise of using centrality measures in ASD gene discovery? Centrality measures help identify the most "important" or influential genes within complex biological networks, such as protein-protein interaction (PPI) networks. The core premise is that genes causing a complex polygenic disorder like Autism Spectrum Disorder (ASD) are not isolated; they often work in concert within key biological pathways. By leveraging network centrality, machine learning models can prioritize genes that occupy crucial positions in these networks, moving beyond simple gene-variant lists to understanding their functional relationships [26] [30].
Q2: How does the forecASD model incorporate network centrality? The forecASD model utilizes a brain-specific gene network that integrates various data types, including gene co-expression and PPI evidence. From this weighted network, it extracts multiple network topology features to characterize each gene. These centrality and importance measures include [30]:
- Node Centrality: Degree, betweenness, and eigenvector centralities.
- Algorithmic Measures: PageRank, which counts the number and quality of links to a node.
- Module Analysis: Features like hub score and coreness to identify key modules within the network. These features are then used as input for prediction, allowing the model to learn which network positions are most associated with known ASD risk genes [30].
Q3: What specific problem does the Stacking-SMOTE model address in this field? The Stacking-SMOTE model directly tackles the critical issue of imbalanced datasets in ASD gene prediction. In resources like the SFARI database, the number of known ASD genes (the minority class) is vastly outnumbered by genes not associated with ASD (the majority class). This imbalance can cause machine learning models to become biased and perform poorly in identifying the very genes researchers want to find. Stacking-SMOTE solves this by generating synthetic data for the minority class to create a balanced dataset for training, thereby reducing model bias and overfitting [31].
Q4: My model's performance is poor. How can I troubleshoot data-related issues? Poor performance often stems from problems with the training data. Focus on these areas:
- Check for Class Imbalance: Evaluate the distribution of your positive (ASD risk genes) and negative (non-ASD genes) classes. If there is a severe imbalance, employ resampling techniques like SMOTE, as used in the Stacking-SMOTE model, to rebalance the dataset [31].
- Validate Feature Quality: Ensure the network centrality features are calculated correctly on a relevant biological network. The predictive power of models like forecASD relies heavily on the quality of the underlying PPI and brain-specific co-expression data [30].
- Verify Data Labels: Use high-confidence gene sets for training. For example, many models use SFARI Gene categories (1, 2, 3, and syndromic) as true positives and genes associated with non-mental health diseases as true negatives to ensure label reliability [30].
Q5: What are the key validation steps for a new ASD gene prediction? Robust validation is essential to build confidence in your predictions. A standard protocol includes:
- Cross-Validation: Use k-fold cross-validation on your training set to ensure the model is not overfitting.
- Independent Test Sets: Hold out a portion of known high-confidence ASD genes (e.g., from SFARI) to test the final model's performance.
- Enrichment Analysis: Test if the top-ranked predicted genes are statistically enriched for known ASD genes from independent sources or for genes involved in biological pathways previously linked to ASD (e.g., chromatin remodeling, synaptic function) [30].
- Literature & Database Verification: Check predicted genes against recent publications and databases like SFARI to see if they have been independently implicated after your model was trained [26] [30].
Troubleshooting Guides
Issue 1: Handling Imbalanced Datasets in ASD Gene Prediction
Problem: Your classifier shows high overall accuracy but fails to identify any novel ASD risk genes because it is biased toward the majority class (non-ASD genes).
Solution: Implement the Synthetic Minority Oversampling Technique (SMOTE).
Protocol:
- Identify Minority Class: Define your positive class (e.g., known ASD genes from SFARI).
- Apply SMOTE: For each instance in the minority class, SMOTE calculates the k-nearest neighbors. It then creates synthetic examples along the line segments joining the minority class instance and its neighbors [31].
- Generate Synthetic Data: This process creates new, synthetic data points for the minority class rather than simply duplicating existing ones, which helps reduce overfitting.
- Re-train Model: Train your model on the newly balanced dataset. The Stacking-SMOTE model demonstrated that this approach can achieve high accuracy (approximately 95.5%) in predicting ASD genes [31].
Issue 2: Integrating and Validating Centrality Features
Problem: The network centrality features you've computed do not improve your model's predictive power for identifying ASD genes.
Solution: Ensure the biological network and centrality measures are contextually relevant to ASD neurobiology.
Protocol:
- Network Selection:
- Feature Extraction: Calculate a diverse set of network features for each gene, as done in forecASD [30]:
- Centrality Measures: Degree, betweenness, eigenvector centrality.
- Influence Measures: PageRank, hub score.
- Topological Measures: Coreness.
- Feature Imputation: For genes present in the expression data but missing from the PPI network, impute their network features using an algorithm like k-Nearest Neighbors [30].
- Biological Validation: Perform enrichment analysis on the genes ranked highly by your model. Check if they are involved in pathways previously associated with ASD, such as synaptic signaling, chromatin remodeling, or the immune system, to confirm the biological relevance of your features [26] [30].
Experimental Protocols & Data
Quantitative Model Performance Comparison
The following table summarizes the performance and key characteristics of the discussed models as reported in the literature.
| Model Name | Core Methodology | Key Technical Features | Reported Performance |
|---|---|---|---|
| forecASD [30] | Network-based ensemble classifier | Brain-specific spatiotemporal co-expression, Protein-Protein Interaction (PPI) networks, PageRank & other centrality measures, Gene-level constraint (pLI) | High predictive power; top-ranked genes enriched for known ASD genes and relevant pathways (e.g., chromatin remodeling). |
| Stacking-SMOTE [31] | Hybrid stacking ensemble with SMOTE | Hybrid Gene Similarity (HGS), Synthetic Minority Oversampling (SMOTE), Gradient Boosting-based Random Forest (GBBRF) classifier | ~95.5% accuracy on SFARI gene database; effective handling of imbalanced data. |
| Game Theoretic Centrality [26] | Coalitional Game Theory (CGT) with biological networks | Shapley value to evaluate gene synergy in networks, Incorporation of prior biological knowledge from PPI networks | Successfully prioritized immune system pathways (e.g., HLA genes) and known ASD genes; offers a novel centrality concept. |
| mantis-ml (NDD) [32] | Semi-supervised machine learning | Integration of single-cell RNA-seq data with 300+ features (intolerance, PPI), Inheritance-specific model training | High predictive power (AUCs: 0.84-0.95); top genes were 45-180x more likely to have literature support. |
Detailed Methodology: Stacking-SMOTE Model Workflow
This protocol outlines the step-by-step process for implementing the Stacking-SMOTE model for ASD gene prediction [31].
Workflow Diagram
Step-by-Step Protocol:
- Data Acquisition & Preprocessing:
- Source: Download all candidate ASD genes from the Simons Foundation Autism Research Initiative (SFARI) database (https://gene.sfari.org/).
- Filtering: Utilize genes from categories 1, 2, 3, and 4 for analysis.
- Annotation: Annotate all candidate genes with Gene Ontology (GO) terms, focusing specifically on the Biological Process (BP) branch for relevant functional information.
Gene Similarity Matrix Construction:
- Function: Apply a Hybrid Gene Similarity (HGS) function. This function combines information gain-based methods with graph-based methods to effectively measure the semantic similarity between genes using their GO annotations.
Handling Data Imbalance:
- Technique: Apply the Synthetic Minority Oversampling Technique (SMOTE) to the dataset.
- Action: SMOTE generates synthetic data samples for the minority class (known ASD genes) instead of simply duplicating existing data, which creates a balanced dataset and reduces the risk of overfitting.
Base Model Training:
- Algorithms: Train multiple base machine learning classifiers on the balanced dataset. The Stacking-SMOTE model uses Random Forest (RF), k-Nearest Neighbors (KNN), Support Vector Machine (SVM), and Logistic Regression (LR).
Stacking Ensemble:
- Meta-Classifier: The predictions from the base classifiers (RF, KNN, SVM, LR) are used as input features to train a meta-classifier.
- Meta-Algorithm: The model uses a novel Gradient Boosting-based Random Forest (GBBRF) as the meta-classifier, which combines the strengths of both boosting and Random Forest to form a robust final prediction model.
Evaluation & Prediction:
- The model is evaluated via cross-validation against the SFARI database and other candidate ASD gene sets.
- The final stacked model is used to output predictions and rank novel ASD risk genes.
Detailed Methodology: forecASD Model Workflow
This protocol describes the process for building a network-based model like forecASD that leverages centrality measures [30].
Workflow Diagram
Step-by-Step Protocol:
- Curate Labeled Gene Set:
- True Positives (TP): Compile a set of high-confidence ASD genes. This typically includes SFARI Gene categories 1 and 2, along with genes from major sequencing studies.
- True Negatives (TN): Compile a set of genes not associated with ASD, such as those linked to non-mental health diseases from resources like OMIM.
Construct a Weighted Functional Network:
- Data Sources:
- Gene Expression: Obtain spatiotemporal RNA-Seq data from the BrainSpan Atlas of the Developing Human Brain.
- Protein Interactions: Obtain PPI data from a database like InWeb.
- Network Construction: For gene pairs with PPIs, calculate their connection weight based on their Fischer z-transformed Pearson correlation coefficient derived from their BrainSpan expression profiles across all brain regions and developmental stages.
- Data Sources:
Feature Extraction:
- Network Features: For each gene in the network, calculate a suite of network features using a tool like the
igraphpackage in R. These include:- Centrality Measures: Degree, betweenness, eigenvector centrality.
- Influence Algorithms: PageRank.
- Topological Measures: Coreness, hub score.
- Constraint Features: Incorporate gene-level constraint metrics from large-scale sequencing projects (e.g., gnomAD), such as pLI scores and Z-scores for LoF, missense, and synonymous variants.
- Expression Features: Use the log-transformed gene expression values from BrainSpan directly as features.
- Network Features: For each gene in the network, calculate a suite of network features using a tool like the
Model Training and Prediction:
- Train a machine learning classifier (e.g., SVM, Random Forest) using the assembled features to distinguish between TP and TN genes.
- The trained model can then score and rank all other genes in the genome based on their predicted probability of being an ASD risk gene.
Biological Validation:
- Perform Gene Ontology (GO) enrichment analysis on the top-ranked genes to identify overrepresented biological processes (e.g., neuronal signaling, chromatin remodeling).
- Check for enrichment in independent sets of ASD risk genes not used in training.
- Examine differential expression evidence of top-ranked genes in ASD brain tissues.
The Scientist's Toolkit: Research Reagent Solutions
| Research Reagent / Resource | Function in Experiment | Key Details / Application |
|---|---|---|
| SFARI Gene Database | Provides curated lists of ASD candidate genes for model training and validation. | Categories 1, 2, 3, and syndromic genes are often used as high-confidence positive labels; essential for benchmarking [31] [30]. |
| BrainSpan Atlas | Source of spatiotemporal human brain gene expression data (RNA-Seq). | Used to build brain-specific co-expression networks and as direct input features; captures developmental dynamics critical to ASD [30]. |
| InWeb PPI Network | Provides a catalog of protein-protein interactions. | Integrated with expression data to build a functionally weighted gene-gene interaction network for centrality analysis [30]. |
| Gene Ontology (GO) | A hierarchical database of gene functional annotations. | Used to calculate semantic similarity between genes (e.g., HGS function) and for post-prediction enrichment analysis [31]. |
| ExAC/gnomAD | Database of genetic variation from a large population. | Source for gene-level constraint metrics (e.g., pLI, missense Z-score), which are key features for predicting gene intolerance to mutation [30]. |
| SMOTE | An algorithm to generate synthetic samples for the minority class in a dataset. | Critical for resolving class imbalance in ASD gene datasets, improving model ability to identify true risk genes [31]. |
| Coalitional Game Theory (CGT) | A mathematical framework to evaluate the marginal contribution of a player (gene) in a coalition. | Used in Game Theoretic Centrality to rank genes by their synergistic influence within a biological network, incorporating prior knowledge [26]. |
Frequently Asked Questions
Q1: Why is my prioritized gene list dominated by general cellular housekeeping genes, and how can I make it more specific to ASD neurobiology?
This is a common issue when the Protein-Protein Interaction (PPI) network is not sufficiently contextualized. A gene with high betweenness centrality might be a general hub, not necessarily specific to brain function or ASD.
- Solution: Integrate brain-specific expression data directly into your gene prioritization. After calculating centrality measures, filter your gene list based on expression levels in relevant brain regions and developmental periods. For example, you can cross-reference your list with data from the BEST (Brain Expression Spatio-Temporal) web server, which provides comprehensive human brain spatio-temporal expression patterns [33]. Retain only genes expressed above a certain threshold (e.g., TPM > 1) in brain regions implicated in ASD, such as the cortex, during critical prenatal or early postnatal developmental windows [34] [35].
Q2: After integrating spatiotemporal data, my gene list becomes too small. How do I balance specificity with statistical power?
Overly stringent spatiotemporal filters can lead to a drastic reduction in candidate genes.
- Solution: Employ a tiered filtering approach instead of a single hard cutoff.
- Tier 1 (High Confidence): Genes with high centrality AND high expression in key brain regions/periods.
- Tier 2 (Medium Confidence): Genes with high centrality OR high expression, plus supporting evidence from other sources (e.g., SFARI gene score, literature).
- Use the gene set from Tier 1 for pathway enrichment analysis to understand core mechanisms, and use the larger set from Tier 2 for hypothesis generation and further validation.
Q3: What are the best public resources to obtain brain spatiotemporal expression data for my candidate genes?
Several high-quality, publicly available resources can be used.
- Primary Resources:
- BEST (Brain Expression Spatio-Temporal pattern web server): A dedicated tool for comprehensive human brain spatio-temporal expression pattern analysis. It allows you to input a gene list and visualize expression quantifications across brain regions and developmental stages [33].
- BrainSpan Atlas of the Developing Human Brain: Provides transcriptome data from post-mortem human brains across multiple developmental periods and brain structures [33] [35].
- Allen Brain Atlas: Includes data on gene expression in the adult and developing human brain [33].
Q4: How can I visually communicate the logic of combining centrality with spatiotemporal filtering in my research paper?
A clear workflow diagram is the most effective way. The diagram below illustrates the step-by-step process, from data integration to final candidate prioritization.
Troubleshooting Guides
Problem: Weak or No Enrichment in Biologically Relevant Pathways
Potential Causes and Solutions:
- Cause 1: Noisy or Non-Biological Centrality. High-centrality genes might be connecting disparate network regions without forming a coherent biological module.
- Action: Before spatiotemporal filtering, perform an initial pathway enrichment analysis on the top-centrality genes. If results are nonspecific (e.g., enriched for "metabolic processes"), your PPI network may lack context. Consider using a brain-specific PPI network if available.
- Cause 2: Incorrect Spatiotemporal Context. You might be looking at the wrong brain region or developmental period.
- Action: Consult the literature to identify the brain regions (e.g., prefrontal cortex, striatum) and developmental periods (e.g., mid-fetal, early childhood) most strongly implicated in ASD [34] [35]. Use the BEST server to systematically test different spatiotemporal categories for enrichment [33].
- Cause 3: Over-Filtering. The combined filters may be too strict, removing genuine signals.
- Action: Widen the spatiotemporal criteria gradually. Instead of requiring high expression in one specific region, consider expression in any of several cortical regions. Re-run the enrichment analysis at each step to find the optimal balance.
Problem: Inconsistent Results Between Different PPI Databases or Expression Atlases
Potential Causes and Solutions:
- Cause 1: Differences in Database Curation. PPI databases have varying curation standards and experimental sources. Expression atlases may use different sequencing platforms, normalization methods, and sample processing protocols.
- Action: This is a known challenge. The best practice is to perform your analysis on two or three consensus datasets.
- For PPI, use a consolidated source like IMEx, as done in the referenced study [25].
- For expression, use a tool like BEST, which integrates data from multiple sources (BrainSpan, GTEx, Allen Brain Atlas) to provide a more robust view [33].
- Report the results that are consistent across multiple data sources as your most reliable findings.
- Action: This is a known challenge. The best practice is to perform your analysis on two or three consensus datasets.
Experimental Protocols & Data
Table 1: Key Quantitative Metrics from a Systems Biology Study on ASD Genes [25]
This table summarizes the core data from a foundational study that built a PPI network from SFARI genes, which you can use as a benchmark for your own experiments.
| Metric | Description | Value in SFARI-Based Network |
|---|---|---|
| Network Nodes | Total proteins in the PPI network. | 12,598 |
| Network Edges | Total physical interactions between proteins. | 286,266 |
| SFARI Gene Coverage | Percentage of high-confidence (Score 1) SFARI genes included in the network. | 96.5% |
| Brain-Expressed Nodes | Percentage of nodes in the network expressed in at least one brain area. | 94.3% |
| Key Centrality Metric | The primary topological measure used for gene prioritization. | Betweenness Centrality |
Protocol 1: Building a PPI Network and Calculating Centrality for ASD Candidate Genes
This protocol outlines the methodology for the initial centrality analysis [25].
- Gene List Curation: Compile a starting list of genes. This can be from databases like SFARI (e.g., scores 1 and 2) or from your own genetic studies (e.g., genes within CNVs of unknown significance).
- Network Generation: Query a consolidated PPI database like IMEx to retrieve both the initial gene products and their first-order interactors.
- Network Construction: Build an undirected PPI network where nodes represent proteins and edges represent physical interactions. Tools like NetworkX in Python can be used for this [36].
- Topological Analysis: Calculate network centrality measures for each node. Betweenness centrality is highly effective, as it identifies nodes that act as bridges connecting different parts of the network [25] [37].
- Prioritization: Rank all genes in the network by their betweenness centrality score in descending order. This is your initial prioritized list.
Protocol 2: Integrating Brain Spatiotemporal Expression Data
This protocol describes how to add a neurobiological context to your computationally prioritized gene list [33] [35].
- Data Source Selection: Access a spatiotemporal brain expression resource. The BEST web server (http://best.psych.ac.cn) is recommended for its user-friendly interface and integrated data [33].
- Input Preparation: Input your ranked gene list into the BEST server. The server can accept a simple list or a list with p-values.
- Spatiotemporal Analysis:
- Generate expression heatmaps to view the expression levels of your gene set across various brain regions and developmental periods.
- Perform cell-type-specific gene set enrichment to see if your genes are overrepresented in specific neural cell types (e.g., neurons, microglia, oligodendrocytes).
- Co-expression Cluster Analysis:
- Use the BEST server to perform co-expression gene cluster enrichment analysis. This identifies if your candidate genes are part of tightly co-regulated gene modules, which often correspond to functional biological pathways.
- Construct co-expression networks to visualize the relationships between your candidate genes within these modules.
- Filtering and Validation: Filter your initial centrality-ranked list, giving priority to genes that are both central and show high expression in ASD-relevant brain spatiotemporal contexts or belong to relevant co-expression clusters.
Table 2: Essential Research Reagent Solutions
This table lists key datasets and software tools that form the essential "reagents" for conducting these analyses.
| Resource Name | Type | Primary Function in Analysis | Access Link / Reference |
|---|---|---|---|
| IMEx Database | Protein-Protein Interaction Data | Provides curated, non-redundant physical protein interactions to build the foundational network. | https://www.imexconsortium.org [25] |
| BEST Web Server | Brain Spatiotemporal Expression Tool | Analyzes and visualizes gene expression patterns across human brain regions and developmental stages. | http://best.psych.ac.cn [33] |
| NetworkX (Python) | Software Library | Performs network construction, calculation of centrality measures, and other graph theory analyses. | https://networkx.org [36] |
| SFARI Gene Database | Gene Annotation Database | Provides a curated list of genes associated with ASD, used for benchmarking and initial gene selection. | https://gene.sfari.org [25] |
| BrainSpan Atlas | Transcriptomics Data | Serves as a key data source within BEST for developmental transcriptome information in the human brain. | http://www.brainspan.org [33] [35] |
Diagram: Conceptual Relationship Between Centrality and Spatiotemporal Expression
The following diagram illustrates the core hypothesis behind this feature engineering approach: that the most robust ASD candidate genes lie at the intersection of high network centrality and high brain-relevant spatiotemporal expression.
Frequently Asked Questions (FAQs)
Q1: What is the core principle behind using network centrality for ASD gene prioritization? Network centrality operates on the "guilt-by-association" principle. It posits that genes causing the same disease are more likely to interact with each other or reside in the same network neighborhood. By mapping known and candidate ASD genes onto a Protein-Protein Interaction (PPI) network, centrality measures can identify and rank genes based on their topological importance, prioritizing those that occupy strategically important positions for further experimental validation [25].
Q2: My candidate gene list contains many genes of unknown significance. How can forecASD help prioritize them? forecASD is specifically designed to handle noisy datasets, including those with Variants of Unknown Significance (VUS). By mapping your candidate gene list onto the pre-compiled PPI network (e.g., derived from SFARI and IMEx), the tool ranks them based on their betweenness centrality. Genes with higher scores are more likely to be true positives. One study using this method successfully prioritized genes within copy number variants, revealing significant enrichment in pathways like ubiquitin-mediated proteolysis [25].
Q3: How does the Game Theoretic Centrality used in forecASD differ from traditional centrality measures?
Game Theoretic Centrality, based on Shapley value from coalitional game theory, evaluates a gene's synergistic influence within a network. Unlike traditional measures like degree centrality, it considers the combinatorial effect of groups of variants. It preferentially ranks genes that are connected to a large number of genes that themselves have few neighbors, identifying influential players that might be missed by other methods. Studies show it identifies a distinct set of genes (e.g., ATP6AP1, GUCY2F) with lower overlap (10-20%) with genes ranked by degree or betweenness centrality [26] [1].
Q4: I only want to use high-confidence, experimentally validated physical interactions from STRING. How can I configure forecASD? Within the forecASD data settings, you can select specific evidence channels. To use only direct experimental data, you would deselect all evidence sources except for "Experiments". STRING integrates experimental data from sources like BIND, DIP, GRID, HPRD, IntAct, MINT, and PID [38] [39]. This ensures the PPI network is built from physical interactions documented in these databases.
Q5: A gene highly ranked by forecASD has no prior link to ASD in the literature. How should I interpret this result?
This is a key strength of the predictive method. A high rank indicates that the gene is topologically important in a network strongly enriched for validated ASD genes. This can reveal novel, biologically plausible candidates. For example, systems biology approaches have prioritized genes like CDC5L, RYBP, and MEOX2 as novel ASD candidates, while game theoretic methods identified GUCA1C and PDE4DIP, which are involved in pathways linked to neurodevelopment [25] [1].
Troubleshooting Guides
Issue 1: Low Overlap Between Prioritized Genes and Known ASD Genes
Problem: The list of genes prioritized by forecASD shows a low overlap with known, high-confidence ASD genes from databases like SFARI.
Solution:
- Verify Network Specificity: Ensure your background PPI network is sufficiently enriched for ASD biology. The network should be built from core ASD genes (e.g., SFARI Score 1 and 2) and their first interactors. A well-constructed network should contain >95% of known high-confidence SFARI genes, a significantly higher percentage than expected by randomly selecting an equal number of genes from the genome [25].
- Adjust Centrality Measure: Different centrality measures capture different aspects of network importance. Consider using Game Theoretic Centrality, which has been shown to identify a novel set of ASD-associated genes (including immune-related genes like
HLA-A,HLA-B,HLA-G) that have lower overlap with other methods but are biologically validated [26] [1]. - Check for Expression Relevance: Confirm that the top-ranked genes are expressed in the brain. Cross-reference your results with expression data from resources like the Human Protein Atlas; a valid network should have over 94% of its nodes expressed in at least one brain area [25].
Issue 2: Handling Genes Not Found in the STRING PPI Network
Problem: Some candidate genes, particularly non-coding genes or pseudogenes, are missing from the STRING network and are therefore excluded from the analysis.
Explanation: STRING is a locus-based database that typically stores a single protein-coding transcript per gene locus and relies on available protein product annotations [38] [39]. This means poorly annotated genes or pseudogenes are often absent.
Solution:
- Pre-filter Input Genes: Before analysis, filter your candidate gene list against the list of proteins available in STRING for your organism of interest (e.g., human).
- Leverage Orthology: For a gene not found in human STRING, check if orthologs exist in model organisms. STRING uses precomputed orthology to transfer interaction evidence across species, which might provide a proxy network [40].
- Acknowledge the Limitation: This is a known constraint of PPI-based methods. The forecASD framework using Game Theoretic Centrality is primarily limited to well-annotated protein-coding genes, as noted in the original research [1].
Experimental Protocol: Validating Centrality Measures for ASD Gene Discovery
The following protocol outlines the key methodology for using a systems biology approach to prioritize and validate ASD candidate genes, as implemented in tools like forecASD.
Constructing the Autism-Related Protein-Protein Interaction (PPI) Network
Objective: To build a comprehensive, ASD-enriched PPI network that will serve as the scaffold for centrality analysis.
Materials & Reagents:
- Source of ASD Genes: Simons Foundation Autism Research Initiative (SFARI) Gene database (https://gene.sfari.org/).
- PPI Data: International Molecular Exchange Consortium (IMEx) database or the STRING database (https://string-db.org/).
- Computational Environment: A computing environment with programming capabilities (e.g., R, Python) and access to network analysis libraries (e.g., igraph, NetworkX).
Procedure:
- Query the SFARI database to obtain a high-confidence seed list of ASD-associated genes. A typical query focuses on non-syndromic genes with SFARI Score 1 ("high confidence") and Score 2 ("strong candidate").
- Retrieve protein interactions for the seed list from a PPI database. Using the IMEx or STRING API, fetch the first-order interactors (direct partners) of all seed genes.
- Merge and assemble the network. Combine the seed genes and their first-order interactors to create a network where nodes represent proteins and edges represent physical interactions.
- Validate network enrichment. Confirm that the constructed network is significantly enriched for known ASD genes compared to a randomly generated network of the same size. This can be tested with a one-sample t-test (p-value < 2.2 × 10−16) [25].
Calculating Centrality Measures for Gene Prioritization
Objective: To compute topological scores for every gene in the network to identify key players.
Procedure:
- Represent the network as a graph object within your chosen computational library.
- Calculate centrality metrics. Compute the betweenness centrality for every node. Optionally, calculate other metrics for comparison (e.g., degree centrality, closeness centrality).
- Rank the genes. Create a prioritized list by sorting all genes in descending order of their betweenness centrality score.
Table: Top 5 Genes by Betweenness Centrality in an Example SFARI-Based Network
| Gene Symbol | SFARI Score | Betweenness Centrality | Relative Betweenness (%) | Known OMIM Phenotype |
|---|---|---|---|---|
| ESR1 | 0.0441 | 100.00 | ||
| LRRK2 | 0.0349 | 79.14 | #607060 (Parkinson's) | |
| APP | 0.0240 | 54.42 | #104300 (Alzheimer's) | |
| JUN | 0.0200 | 45.35 | ||
| CFTR | 0.0189 | 42.86 | #602421 (Cystic Fibrosis) |
Data adapted from a systems biology study of ASD [25].
Functional Validation via Pathway Enrichment Analysis
Objective: To biologically validate the top-ranked genes by identifying the pathways they regulate.
Materials & Reagents:
- Pathway Analysis Tool: Reactome Pathway Browser (https://reactome.org) or similar software (e.g., g:Profiler, Enrichr).
Procedure:
- Select the top-ranked genes from the list generated in Step 2 (e.g., the top 100).
- Submit the gene list to the pathway analysis tool. Use the organism setting as "Homo sapiens."
- Run over-representation analysis (ORA). This typically uses a statistical test like the Fisher exact test, with multiple-testing correction (e.g., Benjamini-Hochberg FDR).
- Interpret the results. Significant pathways (FDR < 0.05) provide biological context. Validated results often include pathways like "Immune System," "Cytokine Signaling," and "Olfactory Signaling", which have been previously implicated in ASD [1] [25].
Table: Key Resources for forecASD and Related ASD Gene Discovery Workflows
| Resource Name | Type | Function in Analysis | Reference/Link |
|---|---|---|---|
| SFARI Gene | Database | Primary source for high-confidence ASD seed genes for network construction. | gene.sfari.org |
| STRING | Database | Provides comprehensive PPI data, integrating known and predicted interactions from multiple evidence channels. | string-db.org [40] |
| IMEx Consortium | Database | Curated repository of experimentally verified molecular interactions to build high-quality PPI networks. | imexconsortium.org [25] |
| Reactome | Database | Used for pathway over-representation analysis to biologically validate prioritized gene lists. | reactome.org [1] |
| igraph/NetworkX | Software Library | Standard libraries for network analysis and calculating centrality measures in R and Python, respectively. | - |
| Human Protein Atlas | Database | Validates brain expression of prioritized candidate genes, adding supporting evidence for relevance to ASD. | proteinatlas.org [25] |
Workflow and Pathway Visualization
forecASD Gene Prioritization Workflow
Game Theoretic vs. Traditional Centrality
Frequently Asked Questions (FAQs)
Q1: What are centrality measures and why are they used in ASD gene discovery? Centrality measures are graph-based metrics that quantify the importance of nodes (genes) within biological networks like protein-protein interaction (PPI) networks. They help prioritize candidate ASD risk genes by identifying genes that occupy strategically important positions in biological networks, with the hypothesis that these genes are more likely to be functionally important in ASD pathophysiology [9] [25].
Q2: Which centrality measures are most commonly used in ASD research? Several centrality measures are commonly employed, each capturing different aspects of network importance:
- Betweenness Centrality: Measures how often a node lies on shortest paths between other nodes [25]
- Degree Centrality: Counts the number of direct connections a node has [9]
- Closeness Centrality: Measures how quickly a node can reach all other nodes [9]
- Eigenvector Centrality: Measures a node's influence based on the importance of its neighbors [9] Recent studies have also introduced more advanced measures like Game Theoretic Centrality based on Shapley value, which considers the synergistic influence of genes in networks [1].
Q3: Can I rely solely on centrality measures to identify causal ASD genes? No. While centrality measures can effectively prioritize candidate genes, they should not be used as a substitute for causal inference. Studies have shown that the correlation between high centrality and actual causal influence can be weak. Centrality measures are excellent for generating hypotheses and prioritizing candidates, but functional validation and causal inference methods are necessary to establish true biological mechanisms [41].
Q4: What are the main limitations of using centrality-based approaches? Key limitations include:
- Inability to distinguish between causal and reactive relationships in the network
- Dependence on the completeness and quality of the underlying PPI network
- Potential bias toward well-studied genes
- Lack of tissue and developmental stage specificity in general PPI networks [30] [41]
- Difficulty interpreting results in highly disconnected or complete graphs [9]
Q5: How can I validate centrality-based predictions experimentally? Multiple validation strategies should be employed:
- Cross-reference predictions with established ASD gene databases (e.g., SFARI Gene)
- Conduct pathway enrichment analysis to identify biologically relevant processes
- Perform functional studies in model systems
- Integrate with gene expression data from relevant tissues and developmental stages
- Compare with differential expression evidence from ASD brains [25] [30]
Troubleshooting Guides
Issue: Poor Performance in Distinguishing ASD Risk Genes
Problem: Your centrality-based classifier fails to distinguish known ASD genes from non-ASD genes effectively.
Solution:
- Verify Network Quality: Ensure your PPI network is comprehensive and specific. Use multiple databases (STRING, IMEx) and consider brain-specific networks [30].
- Combine Multiple Centrality Measures: Implement a hybrid approach using multiple centrality measures rather than relying on a single metric [9].
- Integrate Additional Features: Combine centrality with other genomic features:
Experimental Protocol: Multi-Feature Integration
Issue: Handling Network Topology Challenges
Problem: Your network has problematic topology (e.g., too many disconnected components or overly dense connections) affecting centrality calculations.
Solution:
- Preprocess Network: Remove isolated nodes and small connected components (e.g., components with <10 nodes) [9]
- Apply Thresholding: Use confidence scores for edges in PPI networks to reduce false positives
- Normalize Centrality Scores: Implement normalized centrality measures that account for network size and density
- Consider Alternative Networks: If standard PPI networks perform poorly, construct brain-specific or spatiotemporal co-expression networks [30]
Experimental Protocol: Network Preprocessing
Issue: Integration with Machine Learning Classifiers
Problem: Integrating centrality measures with machine learning pipelines yields suboptimal predictions.
Solution:
- Feature Engineering: Create a comprehensive feature matrix combining:
- Multiple centrality measures (degree, betweenness, closeness, eigenvector)
- Clustering coefficients
- Network module properties [9]
- Hybrid Modeling: Implement advanced architectures like Graph Convolutional Networks (GCNs) combined with traditional classifiers (e.g., logistic regression) [9]
- Cross-Validation: Use rigorous cross-validation strategies accounting for network structure
- Class Imbalance Handling: Apply appropriate sampling techniques or weighted loss functions for imbalanced gene sets
Implementation Code Snippet:
Comparative Analysis of Centrality Measures
Table 1: Centrality Measures for ASD Gene Discovery
| Centrality Measure | Mathematical Definition | Strengths | Limitations | Validation in ASD |
|---|---|---|---|---|
| Betweenness Centrality | C_B(v) = Σ σ_st(v)/σ_st where σst is total shortest paths between s and t, σst(v) passes through v [9] |
Identifies bottleneck genes; Good for biological networks | Computationally intensive for large networks | Validated in SFARI gene prioritization [25] |
| Degree Centrality | C_D(v) = deg(v)/(N-1) where deg(v) is number of connections, N is total nodes [9] |
Simple, intuitive; Fast computation | Only local information; Biased toward highly studied genes | Often used in initial filtering steps [9] |
| Closeness Centrality | C_C(v) = 1/Σ d(v,j) where d(v,j) is shortest path distance to node j [9] |
Identifies genes that can spread information quickly | Sensitive to disconnected components | Used in hybrid approaches [9] |
| Eigenvector Centrality | C_E(v) = (1/λ) Σ A_{iv} C_E(i) where λ is largest eigenvalue, A is adjacency matrix [9] |
Considers neighbor importance; Good for influence | Biased toward dense regions | Correlates with causal influence [41] |
| Game Theoretic Centrality | Based on Shapley value; marginal contribution to coalitions [1] | Captures synergistic effects; Incorporates biological knowledge | Computationally complex; Limited to annotated genes | Identified HLA genes in multiplex families [1] |
The Scientist's Toolkit
Table 2: Essential Research Reagents and Resources
| Resource Type | Specific Examples | Purpose in ASD Gene Discovery | Key Features |
|---|---|---|---|
| PPI Databases | STRING, IMEx, BioGRID | Construct biological networks for centrality analysis | Confidence scores; Multiple evidence channels; Tissue specificity options [9] [25] |
| ASD Gene Databases | SFARI Gene, AUTBASE | Ground truth for training and validation | Expert-curated; Confidence categories; Regular updates [25] [30] |
| Gene Expression Resources | BrainSpan Atlas, GTEx | Spatiotemporal expression features | Developmental trajectories; Brain region specificity [30] |
| Constraint Metrics | gnomAD pLI scores, LOEUF | Gene-level intolerance to variation | Population-based constraint; Helps prioritize functional variants [30] |
| Network Analysis Tools | NetworkX, igraph, Cytoscape | Centrality calculation and visualization | Multiple algorithms; Visualization capabilities; Extensible [9] [30] |
| Machine Learning Frameworks | Scikit-learn, PyTorch, TensorFlow | Building predictive classifiers | GCN implementations; Standard classifiers; Hyperparameter optimization [9] [30] |
Advanced Methodologies
Hybrid Deep Learning Approach
Experimental Protocol: GCN with Logistic Regression
Network Construction:
- Obtain ASD genes from Autism Informatics Portal (≈1,000 genes after cleaning)
- Build PPI network using STRING database
- Result: Network with 979 nodes, 9,505 edges [9]
Feature Extraction:
- Compute multiple centrality measures (degree, betweenness, closeness, eigenvector)
- Calculate clustering coefficients
- Form feature matrix X with dimensions N × 5 (for 5 topological features) [9]
Model Architecture:
- Input Layer: Graph structure (adjacency matrix) and node features
- Graph Convolutional Network: Extracts node embeddings using network structure
- Logistic Regression Layer: Produces probability scores (0-1) for each gene
- Output: Ranked list of potential key regulator genes [9]
Validation:
- Compare with centrality-only methods
- Evaluate using SFARI gene database and EAGLE framework
- Test infection ability using Susceptible-Infected (SI) model [9]
Game Theoretic Centrality Implementation
Experimental Protocol: Shapley Value-Based Prioritization
Data Preparation:
- Whole genome sequences from multiplex autism families
- Protein-protein interaction network from STRING
- Focus on well-annotated protein-coding genes [1]
Coalitional Game Theory Application:
- Define genes as players in a game
- Evaluate marginal contribution of each gene to coalitions
- Calculate Shapley values to quantify synergistic influence [1]
Validation:
- Pathway enrichment analysis (Reactome Pathway Browser)
- Cross-reference with known ASD genes (SFARI)
- Protein-protein interaction with high-confidence candidate genes [1]
Critical Considerations for Experimental Design
Network Selection and Bias: Be aware that PPI networks are biased toward well-studied genes, potentially inflating their centrality measures. Consider using tissue-specific networks (e.g., brain-specific PPIs) or supplementing with co-expression networks derived from relevant tissues and developmental periods [30].
Causal Inference Limitations: Always remember that high centrality indicates strategic network position but does not necessarily imply causal involvement in ASD. Plan functional validation experiments and consider causal inference methods beyond network position analysis [41].
Multi-Omics Integration: For robust predictions, integrate centrality measures with other data types:
- Gene expression patterns across brain development
- Epigenetic regulation data
- Chromatin interaction maps
- constraint metrics [30]
The field continues to evolve with more sophisticated approaches that combine network centrality with machine learning, multi-omics integration, and advanced validation strategies to build more reliable predictive classifiers for ASD risk gene identification.
Troubleshooting Guide: FAQs for Network Analysis in ASD Gene Discovery
FAQ 1: Why does my Protein-Protein Interaction (PPI) network generate an unmanageably large number of nodes, and how can I refine it?
- Problem: Initial PPI networks constructed from ASD candidate genes can yield an excessively large set of nodes (e.g., over 12,000), making biological interpretation difficult [25].
- Solution: Apply a betweenness centrality filter. Rank all nodes by their betweenness centrality score and select the top candidates for downstream analysis. This prioritizes nodes that act as critical connectors in the network. Validate the refined network by checking for significant enrichment of known high-confidence ASD genes (e.g., SFARI Score 1 & 2 genes) compared to randomly generated gene sets [25].
FAQ 2: How can I handle the "noise" from variants of uncertain significance (VUS) in large genomic datasets like SPARK?
- Problem: Copy number variant (CNV) data and other genomic screenings from large cohorts often contain many VUS, complicating the identification of truly pathogenic variants [25].
- Solution: Implement a systems biology prioritization pipeline. Map genes within CNVs or containing VUS onto a pre-constructed, high-confidence PPI network (e.g., a network built from SFARI genes). Then, rank these genes using a topological metric like betweenness centrality. This helps identify VUS-linked genes that occupy strategically important positions within known ASD-related biological networks [25].
FAQ 3: My polygenic risk scores (PRS) perform poorly when applied to a cohort with ancestral diversity. What is the cause and potential solution?
- Problem: PRS derived primarily from genome-wide association studies (GWAS) in European-ancestry populations show attenuated predictive power in non-European populations due to differences in allele frequencies, effect sizes, and linkage disequilibrium (LD) patterns [42].
- Solution: Prioritize the inclusion of ancestrally diverse participants in your discovery GWAS. When analyzing multi-ancestry cohorts, use specialized analytical methods that account for population structure, such as principal component analysis (PCA), and consider cross-ancestry meta-analysis approaches, which can improve the portability and accuracy of genetic risk prediction across populations [42].
FAQ 4: How can I validate the biological relevance of hub-bottleneck genes identified in my network analysis?
- Problem: Computational identification of hub-bottleneck genes (genes with high degree and high betweenness centrality) requires functional validation to confirm their relevance to ASD pathophysiology [43].
- Solution: Integrate differential gene expression evidence from independent ASD transcriptome datasets. After identifying hub-bottlenecks (e.g., EGFR, ACTB, CALM1), query their expression profiles in repositories like the Gene Expression Omnibus (GEO) to confirm they are significantly dysregulated in ASD cases compared to controls [43].
Key Experimental Protocols
Protocol for Constructing and Prioritizing a PPI Network for ASD Gene Discovery
Application: This protocol is used to move from a initial list of ASD-associated genes to a refined set of high-priority candidates, as demonstrated in systems biology studies [25].
Detailed Methodology:
- Seed Gene Collection: Compile a foundational list of high-confidence ASD risk genes. A standard resource is the SFARI Gene database, initially focusing on non-syndromic genes from categories SFARI Score 1 and Score 2 [25].
- Network Expansion: Query these seed genes against a curated PPI database (e.g., IMEx) to retrieve their direct interaction partners. Combine seeds and interactions to construct the initial PPI network [25].
- Topological Analysis: Calculate network topology metrics for every node using tools like Cytoscape with its NetworkAnalyzer plugin. Essential metrics include:
- Gene Prioritization: Rank all genes in the network based on their betweenness centrality scores. Genes with higher scores are considered top candidates for further validation [25].
- Enrichment Validation: Test the specificity of your network by comparing its enrichment for known ASD genes against 1,000 randomly generated gene sets of equal size. A statistically significant enrichment (p-value < 2.2e-16) confirms the network's biological relevance [25].
Protocol for Differential Expression and Hub-Bottleneck Analysis
Application: This protocol integrates gene expression data with network analysis to identify and validate key regulatory genes in ASD, a method used in transcriptomic studies [43].
Detailed Methodology:
- Data Acquisition: Download a relevant ASD gene expression dataset (e.g., GEO accession GSE29691) that includes samples from ASD patients and healthy controls [43].
- Quality Control and DEG Identification:
- Use boxplot analysis to verify that samples are median-centered and comparable.
- Identify Differentially Expressed Genes (DEGs) using analytical tools like GEO2R. Apply significance (adjusted p-value < 0.05) and fold-change thresholds (e.g., 0.5 ≥ FC ≥ 1.5) [43].
- PPI Network Construction and Hub-Bottleneck Identification:
- Input the DEG list into Cytoscape using the STRING plugin to retrieve a interaction network.
- Calculate Degree Centrality (DC) and Betweenness Centrality (BC) for each node.
- Select the top 20% of genes with the highest DC and BC values. The overlap of these two groups are defined as "hub-bottleneck" genes [43].
- Expression Cross-Validation: Merge the hub-bottleneck gene list with the original expression dataset to verify which of these topologically important genes are also significantly differentially expressed [43].
Table 1: Top Hub-Bottleneck Genes from an ASD Transcriptomic PPI Network This table lists genes identified as both hubs (highly connected) and bottlenecks (critical connectors) in a PPI network built from differentially expressed genes in ASD, along with their expression changes [43].
| Gene Symbol | Degree Centrality | Betweenness Centrality | Expression Change in ASD | Fold Change |
|---|---|---|---|---|
| EGFR | 51 | 0.06 | Up | 1.69 |
| MAPK1 | 51 | 0.03 | Down | 1.54 |
| CALM1 | 47 | 0.03 | Down | 2.09 |
| ACTB | 46 | 0.02 | Down | 2.09 |
| JUN | 39 | 0.02 | Up | 1.76 |
| RHOA | 44 | 0.02 | Down | 1.62 |
Table 2: Centrality-Based Prioritization of Novel ASD Candidate Genes from a PPI Network This table shows new candidate genes for ASD identified not by direct mutation, but by their high betweenness centrality in a PPI network constructed from known ASD genes [25].
| Gene Symbol | Betweenness Centrality | SFARI Score (if any) | Expression in Brain (TPM) |
|---|---|---|---|
| CDC5L | High (Prioritized) | Not Assigned | Data Required |
| RYBP | High (Prioritized) | Not Assigned | Data Required |
| MEOX2 | 0.0087 | Not Assigned | 0.68 (Low) |
| CUL3 | 0.0150 | Score 1 | 22.88 (Medium) |
| DISC1 | 0.0169 | Score 2 | 2.50 (Low) |
Signaling Pathway and Workflow Diagrams
PPI Network Analysis Workflow
Hub Gene Connects ASD Pathways
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Resources for ASD Gene Discovery Using Network Centrality
| Resource Name | Type/Format | Primary Function in Research | Key Application in ASD Context |
|---|---|---|---|
| SFARI Gene Database | Online Database | Provides curated lists of ASD-associated genes with confidence scores. | Source of high-confidence "seed genes" for building biologically relevant PPI networks [25]. |
| Cytoscape | Software Platform | Visualizes and analyzes molecular interaction networks. | Core tool for constructing PPI networks, calculating centrality metrics (via NetworkAnalyzer), and identifying hub-bottlenecks [43]. |
| STRING Plugin | Cytoscape App | Retrieves protein-protein interaction data from multiple sources directly within Cytoscape. | Streamlines the process of building a PPI network from a list of candidate genes [43]. |
| BrainSpan Atlas | RNA-Seq Dataset | Provides spatiotemporal gene expression patterns in the developing human brain. | Used as a feature in machine learning models to predict ASD risk genes and validate brain-relevance of candidates [30]. |
| ExAC/gnomAD | Population Genomic Database | Provides gene-level constraint metrics (e.g., pLI, Z-scores). | Helps prioritize genes intolerant to loss-of-function mutations, a key characteristic of many ASD risk genes [30]. |
| Simons Searchlight | Patient Cohort & Registry | A "gene-first" research program for specific genetic neurodevelopmental disorders. | Enables deep phenotyping and research on individuals with specific genetic findings from cohorts like SPARK [44]. |
Navigating the Complexities: Troubleshooting and Optimizing Centrality Applications
Addressing Data Imbalance with Techniques like SMOTE
Frequently Asked Questions (FAQs)
Q1: Why is data imbalance a critical problem in ASD gene discovery research? Data imbalance, where the number of known ASD genes (minority class) is vastly outnumbered by non-ASD or non-causal genes (majority class), causes machine learning models to become biased. They will often achieve high accuracy by simply always predicting the "non-ASD" class, failing to identify the novel ASD risk genes that are of primary research interest. Effectively handling this imbalance is therefore essential for building robust predictive models that can prioritize new candidate genes for validation [31] [45].
Q2: How does the SMOTE technique work to address class imbalance? The Synthetic Minority Over-sampling Technique (SMOTE) generates synthetic data rather than simply duplicating existing minority class instances. It works by selecting examples from the minority class that are close in feature space, drawing a line between them, and creating new synthetic examples at points along that line. This technique effectively increases the number of minority class samples and helps the model learn better decision boundaries, thereby reducing the risk of overfitting associated with simple duplication [31].
Q3: Are there techniques that can be combined with SMOTE for better performance? Yes, hybrid approaches that combine SMOTE with other sampling techniques have shown promise. One such method is SMOTE-RUS, which integrates the SMOTE oversampling technique with Random Undersampling (RUS). RUS randomly removes instances from the majority class. When used together, they can create a more balanced and robust dataset for training, leading to a more powerful gene prediction model [45].
Q4: What is an advanced machine learning framework that uses SMOTE for ASD gene prediction? A state-of-the-art framework is the hybrid Stacking-SMOTE model. This model integrates SMOTE for handling imbalanced data with a sophisticated stacking ensemble classifier. The stacking ensemble combines multiple base classifiers (like Random Forest, k-Nearest Neighbors, and Support Vector Machines) using a gradient boosting-based random forest classifier (GBBRF) as a meta-learner. This integrated approach has been shown to optimize the prediction of ASD genes, achieving high accuracy [31].
Troubleshooting Guides
Problem 1: Model is Biased and Fails to Predict Novel ASD Genes
Symptoms:
- High overall accuracy but zero or very low recall for the ASD gene class.
- The model consistently predicts "non-ASD" for almost all test genes.
Solutions:
- Implement SMOTE: Apply SMOTE to your training dataset to synthetically generate new examples of ASD genes. Do not apply it to your test or validation sets to ensure a realistic performance evaluation.
- Tune Hyperparameters: After applying SMOTE, re-tune your model's hyperparameters. The optimal parameters for the imbalanced original dataset are likely suboptimal for the balanced one.
- Use Hybrid Sampling: If SMOTE alone does not suffice, try a combined approach like SMOTE-RUS, which balances the dataset from both the minority and majority classes [45].
Problem 2: Integrating SMOTE into a Complex Ensemble Learning Pipeline
Symptoms:
- Uncertainty about where to place the SMOTE step within a multi-stage workflow.
- Data leakage between training and validation splits, leading to over-optimistic results.
Solutions:
- Follow a Structured Workflow: Adhere to a proven pipeline structure. The diagram below illustrates the integration of SMOTE within a stacking ensemble model, ensuring it is applied only to the training folds during the cross-validation process.
- Prevent Data Leakage: Always perform the SMOTE oversampling after splitting your data into training and testing sets, and inside the cross-validation loop on the training fold only. This prevents the model from having prior knowledge of the test set.
Problem 3: Choosing the Right Evaluation Metrics for an Imbalanced Dataset
Symptoms:
- Relying solely on accuracy to evaluate model performance.
- Good performance on accuracy but poor performance in actually discovering true ASD genes.
Solutions:
- Avoid Accuracy: Stop using accuracy as the primary metric for imbalanced datasets.
- Adopt Comprehensive Metrics: Use a suite of metrics that are robust to class imbalance. The table below summarizes key metrics and their interpretation in the context of ASD gene discovery.
Table 1: Key Performance Metrics for Imbalanced ASD Gene Classification
| Metric | Interpretation in ASD Gene Discovery Context | Formula |
|---|---|---|
| Area Under the ROC Curve (AUC) | Measures the model's ability to distinguish between ASD and non-ASD genes across all classification thresholds. A value of 0.5 is random, and 1.0 is perfect. | N/A |
| Precision | In the top N genes predicted by the model, what proportion are truly associated with ASD? High precision means fewer false positives. | TP / (TP + FP) |
| Recall (Sensitivity) | Of all the known true ASD genes, what proportion did the model successfully identify? High recall means fewer false negatives. | TP / (TP + FN) |
| F1-Score | The harmonic mean of Precision and Recall. Provides a single score to balance the trade-off between the two. | 2 * (Precision * Recall) / (Precision + Recall) |
Abbreviations: TP = True Positive, FP = False Positive, FN = False Negative. [31]
Experimental Protocols
Protocol 1: Implementing a Basic SMOTE-based Resampling Strategy
Objective: To balance an imbalanced ASD gene dataset using SMOTE for improved model training.
Materials: See "The Scientist's Toolkit" below.
Methodology:
- Data Preparation: Load your labeled gene dataset. Ensure it is split into features (e.g., gene similarity scores, constraint metrics) and a target variable (e.g., ASD gene = 1, non-ASD gene = 0).
- Train-Test Split: Split the dataset into a training set (e.g., 70-80%) and a held-out test set (e.g., 20-30%). Do not apply any resampling to the test set.
- Apply SMOTE: Import the SMOTE algorithm from a library like
imbalanced-learn(Python). Apply SMOTE exclusively to the training data to generate synthetic ASD gene samples until the class distribution is balanced (e.g., 1:1 ratio). - Model Training: Train your chosen classifier (e.g., Random Forest, SVM) on the resampled training data.
- Evaluation: Validate the trained model's performance on the original, untouched test set using the metrics in Table 1.
Protocol 2: Validating a Stacking-SMOTE Ensemble Framework
Objective: To implement and evaluate a high-performance Stacking-SMOTE model for ASD gene prediction, as described in recent literature [31].
Methodology: The entire workflow is visualized in the diagram above. The key steps are:
- Data Preprocessing & Splitting: Follow Steps 1 and 2 from Protocol 1. The use of a hybrid gene similarity function (HGS) that combines information gain and graph-based methods to measure functional similarity between genes is recommended for feature engineering [31].
- Base-Level Model Training:
- On the SMOTE-balanced training data, train multiple diverse base classifiers. Typical choices include Random Forest (RF), Support Vector Machine (SVM), k-Nearest Neighbors (kNN), and Logistic Regression (LR).
- Perform k-fold cross-validation on the balanced training set to generate out-of-fold predictions for each base model.
- Meta-Learner Training:
- The out-of-fold predictions from the base models are used as features to train a meta-learner. The proposed model uses a Gradient Boosting-Based Random Forest (GBBRF) for this role, which enhances predictive performance.
- This step combines the strengths of the individual base models.
- Final Evaluation: The fully trained stacking ensemble (base models + meta-learner) is used to make predictions on the held-out test set for a final, unbiased performance assessment.
The Scientist's Toolkit
Table 2: Essential Research Reagents and Computational Tools for ASD Gene Prediction
| Item Name | Function / Description | Relevance to Experiment |
|---|---|---|
| SFARI Gene Database | A curated database of ASD-associated genes from the Simons Foundation Autism Research Initiative. | Serves as the primary source for labeled positive genes (e.g., categories 1, 2, 3) for model training and validation [31] [30]. |
| Gene Ontology (GO) | A major bioinformatics resource that describes gene functions and relationships across three domains: Biological Process (BP), Molecular Function (MF), and Cellular Component (CC). | Used to calculate functional similarities between genes. The Biological Process (BP) branch is often most relevant for constructing gene similarity matrices [31]. |
| Hybrid Gene Similarity (HGS) | A similarity function that combines information gain-based and graph-based methods. | Used to construct a robust gene functional similarity matrix as input features for the classifier, improving prediction accuracy [31]. |
| BrainSpan Atlas | A spatiotemporal transcriptomic dataset of the developing human brain. | Provides gene expression features across different brain regions and developmental stages, which are highly informative for predicting neurodevelopmental disorder genes [30]. |
| Gene-Level Constraint Metrics (e.g., pLI) | Metrics derived from large population sequencing data (e.g., gnomAD) that quantify a gene's intolerance to loss-of-function mutations. | A high pLI score indicates a gene is intolerant to mutations, a key feature for identifying ASD risk genes. Used as a predictive feature in machine learning models [30]. |
Troubleshooting Guide: Ancestral Bias in Genomic Research
FAQ: Addressing Common Experimental Issues
1. My polygenic risk scores (PRS) perform well in European ancestry populations but poorly in others. What is the root cause and how can I address it?
This performance disparity stems from ascertainment bias in training data. Most genomic databases (like TCGA and GWAS Catalog) are predominantly composed of individuals of European ancestry, leading to models that overfit to this specific population structure [46]. The following table summarizes the extent of this bias in major genomic resources:
| Genomic Resource | Reported European Ancestry Proportion | Primary Consequence |
|---|---|---|
| The GWAS Catalog [46] | ~95% | Severely limits understanding of disease drivers in non-European populations. |
| The Cancer Genome Atlas (TCGA) [46] | Median of 83% (range 49-100%) | Poor generalization of risk predictors for cancer in minority populations. |
| Cell Line Transcriptomic Data [46] | ~95% (Only 5% from individuals of African descent) | Models fail to capture the greater genetic diversity present in African populations. |
Solution: Instead of relying on single-ancestry models, employ equitable machine learning frameworks like PhyloFrame or DisPred that explicitly adjust for ancestral distribution shifts. PhyloFrame integrates functional interaction networks and population genomics data with transcriptomic training data to create ancestry-aware signatures [46]. DisPred uses a deep-learning approach to disentangle ancestry from phenotype-relevant information in its genetic representations, improving performance across populations without needing self-reported ancestry for prediction [47].
2. How can I validate that my centrality measures for ASD gene discovery are not biased by ancestral background?
Standard centrality measures (degree, betweenness) applied to protein-protein interaction (PPI) networks can be biased because the networks themselves are often built from data that under-represent non-European populations [46]. This can cause you to prioritize genes that are central only in a specific ancestral context.
Solution: Supplement standard centrality analysis with game-theoretic centrality and functional validation across diverse populations.
- Game-Theoretic Centrality: This approach, based on the Shapley value, evaluates a gene's synergistic influence in a network. It prioritizes genes that are connected to many partners that themselves have few connections, which can reveal influential genes that might be missed by traditional measures [1]. One study found that only 10-20% of top genes identified by game-theoretic centrality overlapped with those from degree or betweenness centrality [1].
- Cross-Ancestry Enrichment Analysis: Validate your candidate genes by testing their enrichment in pathways using diverse datasets. For example, top genes from a game-theoretic analysis were enriched for immune system pathways and included HLA complex genes (HLA-A, HLA-B, HLA-G, HLA-DRB1), which have been implicated in ASD across multiple studies [1].
3. I have limited access to diverse genomic datasets. What is the minimal viable approach to improve the generalizability of my findings?
You can leverage existing methods and public resources that are designed to work with imbalanced data.
- Utilize Equitable ML Methods: Frameworks like PhyloFrame are designed to correct for ancestral bias even when the training data is unbalanced [46]. They can create robust signatures without requiring a perfectly representative dataset.
- Incorporate Gene-Level Constraint Metrics: Use metrics like the probability of being loss-of-function intolerant (pLI) from resources like the Exome Aggregation Consortium (ExAC). ASD genes affected by de novo mutations tend to be intolerant of variations, and this constraint is a useful feature for prediction that can be applied across ancestries [30].
- Leverage Brain-Specific Networks: Instead of general PPI networks, use brain-specific gene co-expression networks (e.g., from the BrainSpan Atlas) that capture spatiotemporal dynamics relevant to neurodevelopment [30].
Experimental Protocol: Implementing an Ancestry-Aware Analysis
Objective: To identify ASD risk genes using a network-based approach that mitigates ancestral bias.
Methodology Overview: This protocol integrates a machine learning framework for equitable prediction with a game-theoretic centrality measure for prioritization.
Workflow Diagram
Step-by-Step Instructions:
Data Collection and Pre-processing:
- Obtain gene expression data from a resource like the BrainSpan Atlas (spatiotemporal data of the developing human brain) [30].
- Gather gene-level constraint metrics (e.g., pLI, LoF Z-scores) from public databases like gnomAD.
- If using individual-level genetic data, ensure you have appropriate ethical approvals and ancestry information if available.
Bias Mitigation with Equitable Machine Learning:
- If using PhyloFrame: Input your transcriptomic training data. The method will integrate population genomics data and functional interaction networks to output an ancestry-adjusted signature of disease [46].
- If using DisPred: Input genotype dosage data. The disentangling autoencoder will separate ancestry-specific representations from phenotype-specific representations, which are then used for prediction [47].
Network Construction and Gene Prioritization:
- Build a weighted gene network using co-expression data (e.g., from BrainSpan) and PPI evidence (e.g., from InWeb) [30].
- Apply a game-theoretic centrality measure to this network to rank genes based on their synergistic influence. This evaluates the marginal contribution of each gene to the overall connectivity [1].
Validation and Interpretation:
- Perform gene ontology (GO) enrichment analysis on the top-ranked genes to identify over-represented biological processes (e.g., neuronal signaling, chromatin remodeling) [30] [1].
- Cross-reference candidates with known ASD gene databases (e.g., SFARI) and check for enrichment in pathways previously linked to ASD (e.g., immune system, ubiquitination) [1].
Research Reagent Solutions
The following table lists key resources for implementing the described methodologies.
| Item / Resource | Function / Application | Key Features / Notes |
|---|---|---|
| BrainSpan Atlas | Provides spatiotemporal gene expression data for the developing human brain. | Essential for building brain-specific co-expression networks; captures dynamic developmental patterns [30]. |
| ExAC/gnomAD | Provides gene-level constraint metrics (e.g., pLI, missense Z-score). | Quantifies a gene's intolerance to variation; a useful feature for prioritizing ASD risk genes [30]. |
| STRING Database | A database of known and predicted Protein-Protein Interactions (PPIs). | Used to build functional interaction networks; includes both physical and functional associations [43]. |
| PhyloFrame | An equitable machine learning method for genomic precision medicine. | Corrects for ancestral bias by integrating functional networks and population genomics data [46]. |
| DisPred | A deep-learning framework for genetic risk prediction. | Disentangles ancestry from phenotype-relevant representations to improve generalizability [47]. |
| igraph R package | A library for network creation, analysis, and visualization. | Can calculate various centrality measures and implement network-based analyses [48]. |
| SFARI Gene Database | A curated database of genes associated with ASD susceptibility. | Serves as a benchmark for validating newly discovered candidate ASD genes [30] [1]. |
Advanced Technical Note: The DisPred Architecture
For researchers seeking to implement the DisPred method, the core of its approach to disentangling ancestry is illustrated in the following workflow. Note that this framework does not require self-reported ancestry information for final predictions, making it suitable for practical applications where such metadata may be unavailable [47].
The foundational premise of network-based biology is that cellular function arises from complex webs of molecular interactions. In the specific context of Autism Spectrum Disorder (ASD) research, protein-protein interaction (PPI) networks have become crucial for prioritizing candidate genes from large-scale genomic studies [25]. Analyses often rely on centrality measures, like betweenness centrality, to identify hub genes that are topologically important and thus potentially biologically significant [25] [43]. However, the power of these predictions is fundamentally constrained by the "Incomplete Interactome Problem"—the fact that current network data is a static, fragmented, and context-blind representation of a dynamic cellular reality. This guide addresses the specific challenges this problem poses for validating centrality measures in ASD gene discovery.
Frequently Asked Questions (FAQs)
1. Why does my network analysis identify different hub genes for ASD than other studies, even when using similar data? This lack of reproducibility often stems from the incomplete and biased nature of interactome maps. Different experimental techniques (e.g., Yeast Two-Hybrid vs. Affinity Purification-Mass Spectrometry) have distinct biases, capturing different subsets of interactions (e.g., stable complexes vs. transient signals) [49]. When you calculate centrality, you are measuring importance within a specific, flawed map. Gaps and technical artifacts in one dataset can lead to the miscalculation of a node's centrality, causing prioritization of different genes across studies [49] [50].
2. My top-ranked candidate gene by betweenness centrality was not expressed in relevant brain tissues. Is the measure invalid? Not necessarily. This discrepancy highlights the problem of context independence. Most canonical PPI networks are amalgamations of interactions from various cell types, tissues, and developmental stages [49]. A protein may be topologically central in a generic network, but biologically irrelevant in a specific context like mid-fetal prefrontal cortex development. The validation of centrality measures must therefore include spatiotemporal expression data from resources like the BrainSpan atlas to ensure biological relevance [30].
3. Why are my network-based predictions for ASD dominated by general, essential genes rather than neurodevelopmental-specific ones? This is a known consequence of methodological bias and network incompleteness. Highly connected "hub" proteins involved in basic cellular processes are more likely to be detected by multiple high-throughput methods, making them appear in current networks with high confidence. In contrast, tissue-specific genes might have fewer, more context-dependent interactions that are missed, artificially lowering their centrality scores [49] [25]. Your results may reflect the current state of the map, not just the biology of ASD.
Troubleshooting Guides
Problem: Static Network Models Lead to Biased Centrality Rankings
Issue: Your analysis uses a single, static PPI network, failing to capture the dynamic nature of molecular interactions during neurodevelopment.
Solution:
- Incorporate Dynamic Data: Move beyond static networks by integrating temporal gene expression data. A key methodology involves building time-specific co-expression networks.
- Experimental Protocol:
- Data Acquisition: Download RNA-Seq data from developmental brain atlases like BrainSpan, which provides gene-level expression values across multiple brain regions and developmental stages [30].
- Network Construction: For a specific spatiotemporal window (e.g., mid-fetal period), calculate pairwise correlations (e.g., Pearson) between gene expression profiles. Construct a weighted network where edges represent co-expression strength [30].
- Centrality Calculation: Perform centrality analysis (e.g., betweenness centrality) on these context-aware networks to identify hubs critical during specific developmental windows [30].
The following workflow outlines this process for creating dynamic, context-aware networks:
Problem: High False Positive Rates in Gene Prioritization
Issue: Your ranked list of candidate genes from network analysis contains many genes that are likely false positives, reducing experimental validation yield.
Solution:
- Apply Constrained Disorder Principle (CDP) Concepts: Recognize that biological variability is not just noise but a functional feature. Prioritize genes that maintain their high centrality ranking across a range of network conditions [49].
- Experimental Protocol: Perturbation-Based Robustness Testing
- Network Perturbation: Systematically introduce noise into your network model. This can involve randomly adding or removing a small percentage of edges, or bootstrapping the original interaction data.
- Re-calculate Centrality: For each perturbed network version, re-compute the betweenness centrality for all nodes.
- Stability Assessment: Rank genes by the stability of their centrality score across these perturbations. Genes that consistently rank high are more reliable candidates than those whose rank is highly variable [49].
The table below summarizes the core limitations of static interactomes and how they impact the validation of centrality measures for ASD gene discovery.
| Limitation | Impact on Centrality Measures (e.g., Betweenness) | Consequence for ASD Gene Discovery |
|---|---|---|
| Static Representation [49] | Fails to capture dynamics of neurodevelopment; centrality becomes an average over many irrelevant states. | May miss genes critical in specific developmental windows. |
| Context Independence [49] | Measures centrality in an aggregate network, not a brain- or neuron-specific one. | Prioritizes generically important genes over neurodevelopmentally relevant ones [30]. |
| Incomplete Coverage [49] | Centrality scores are calculated on a fragmented map (only ~20% of human PPIs known). | Top-ranked genes may be artifacts of the current map's topology, not true biological hubs. |
| Methodological Biases [49] | Y2H favors binary interactions; AP-MS favors stable complexes. Centrality is technique-dependent. | Reduces reproducibility; different methods yield different "top" genes. |
| Neglect of Individual Variation [49] | Uses a "one-size-fits-all" network, ignoring genetic diversity. | Limits utility for personalized medicine and understanding variable ASD expressivity. |
The Scientist's Toolkit: Research Reagent Solutions
Table: Essential Resources for Building and Analyzing ASD PPI Networks
| Research Reagent / Resource | Function in Analysis | Relevance to ASD & Centrality Validation |
|---|---|---|
| STRING / BioGRID / IMEx [49] [25] | Consolidated databases of curated and predicted protein-protein interactions. | Source for constructing the foundational PPI network for centrality calculation. |
| Cytoscape [43] | Open-source platform for network visualization and analysis. | Used to import PPI networks, calculate topological metrics (degree, betweenness), and identify hub-bottlenecks. |
| NetworkAnalyzer (Cytoscape App) [43] | Computes comprehensive network topology parameters. | Directly calculates betweenness centrality and other centralities for nodes in the network. |
| BrainSpan Atlas [30] | A resource for spatiotemporal gene expression data in the developing human brain. | Critical for moving from static to context-aware networks and validating the biological relevance of central genes. |
| CluePedia (Cytoscape App) [43] | Provides enrichment analysis and integrates expression data with networks. | Used to merge hub-bottleneck genes with GEO expression data to check for significant differential expression in ASD. |
| SFARI Gene Database [25] [30] | A curated database of genes associated with ASD risk. | Serves as a benchmark "gold standard" set for testing the performance of centrality-based prediction models. |
Problem: Validating the Biological Relevance of Topological Hubs
Issue: You have a list of genes ranked by betweenness centrality, but need to validate their functional relevance to ASD pathology.
Solution:
- Integrate Multi-Modal Evidence: Corroborate network-based predictions with independent lines of biological evidence.
- Experimental Protocol: Multi-Faceted Validation Pipeline
- Differential Expression Check: Use tools like CluePedia in Cytoscape to overlay your network hub-bottlenecks (e.g., EGFR, ACTB, MAPK1) with gene expression datasets from ASD patients (e.g., from GEO). Confirm they are differentially expressed [43].
- Functional Enrichment Analysis: Perform Gene Ontology (GO) enrichment analysis on your top candidate genes using tools like STRING Enrichment. Look for over-representation in pathways clearly relevant to ASD (e.g., neuronal signaling, axon development, ubiquitination) [25] [30] [43].
- Gene-Level Constraint Metrics: Consult databases like gnomAD for gene-level constraint metrics (e.g., pLI scores). ASD risk genes are often intolerant to loss-of-function mutations, providing supporting genetic evidence [30].
This multi-layered validation strategy is summarized in the following workflow:
This technical support guide provides troubleshooting and methodological support for researchers employing Hybrid Similarity Functions (HGS) and ensemble methods to validate centrality measures in Autism Spectrum Disorder (ASD) gene discovery. The integration of these computational approaches addresses critical challenges in identifying robust ASD risk genes by optimizing model performance and ensuring biological relevance. The protocols herein are designed for scientists and drug development professionals working at the intersection of bioinformatics and complex disorder genetics.
Understanding HGS in ASD Gene Discovery
What is the Hunger Games Search (HGS) algorithm and why is it relevant to ASD gene discovery?
The Hunger Games Search (HGS) is a metaheuristic optimization algorithm inspired by hunger-driven foraging behaviors and competition in biological organisms [51]. In ASD gene discovery, HGS provides a powerful framework for navigating high-dimensional biological data to identify optimal gene subsets and network configurations [52]. The algorithm mimics how hungry animals forage and compete for resources, translating this natural optimization process to computational problem-solving [51].
For ASD research specifically, HGS helps address the polygenic nature of the disorder by efficiently searching through thousands of potential gene interactions to identify the most promising candidates [52]. The algorithm's capability to balance exploration of novel gene associations with exploitation of known ASD-related pathways makes it particularly valuable for validating centrality measures in protein-protein interaction networks [26].
How do ensemble methods improve HGS performance for ASD biomarker identification?
Ensemble methods enhance HGS performance through three primary mechanisms that address core challenges in ASD biomarker identification:
Multi-strategy integration: Combining chaos theory, greedy selection, and vertical crossover operations maintains population diversity while improving convergence rates [52]. This is crucial for avoiding local optima in the complex fitness landscape of ASD genetic architecture.
Phased search coordination: Dynamic coordination of global exploration and local exploitation through distinct search phases prevents premature convergence on spurious gene associations [51].
Hybrid similarity optimization: Integrating biological knowledge from multiple sources (e.g., co-expression networks, protein interactions) creates more robust similarity functions for prioritizing ASD genes [30].
Experimental validations demonstrate that ensemble-enhanced HGS achieves 23.7% average improvement in optimization accuracy compared to single-strategy approaches [51].
Troubleshooting Common Experimental Issues
How can I address premature convergence when applying HGS to ASD gene networks?
Premature convergence typically manifests as repeated identification of the same gene subsets without meaningful improvement in fitness scores. Address this using the following strategies:
Table: Solutions for Premature Convergence in HGS-based ASD Gene Discovery
| Issue | Symptoms | Solution | Expected Outcome |
|---|---|---|---|
| Limited population diversity | Rapid fitness stagnation in early generations | Implement chaotic initialization [52] and enhanced reproduction operators [51] | 15-30% improvement in solution diversity |
| Imbalanced exploration-exploitation | Repeated oscillation between similar solutions | Apply phased position update framework [51] | Better trade-off between novel gene discovery and known pathway validation |
| Insufficient oppositional learning | Inability to escape local optima | Integrate elite dynamic oppositional learning with self-adjusting coefficients [51] | 20% higher likelihood of identifying novel ASD candidates |
Implementation protocol for chaotic initialization:
- Select an appropriate chaotic map (Logistic or Tent maps recommended)
- Replace random values in initial population generation with chaotic sequences
- Validate population distribution across search space dimensions
- Proceed with standard HGS optimization workflow
What are the recommended strategies for handling high-dimensional genomic data in HGS workflows?
High-dimensional genomic data presents challenges including computational overhead and curse of dimensionality. Effective strategies include:
Binary HGS implementation: For feature selection tasks, implement a binary variant (BHGS) using sigmoid transformation [52]. This approach has achieved 92.3% average classification accuracy on UCI genomic datasets.
Multi-stage filtering:
Dimensionality reduction: Employ deep autoencoder neural networks (DAEN) to project high-dimensional data to informative lower-dimensional representations [53].
How can I validate that my HGS-optimized centrality measures are biologically meaningful for ASD?
Biological validation requires multi-faceted approaches beyond computational metrics:
Pathway enrichment analysis: Use Reactome Pathway Browser (reactome.org) to test enrichment of HGS-identified genes in ASD-relevant pathways [1]. Significant pathways include immune system (FDR = 2.15×10⁻¹⁵), synaptic transmission, and chromatin remodeling.
Cross-reference with established ASD databases: Compare your results with:
Experimental prioritization: Focus validation efforts on genes that:
Experimental Protocols & Workflows
Standardized workflow for HGS-based ASD gene discovery
Detailed protocol for multi-strategy HGS implementation
Protocol Title: Implementation of Multi-strategy HGS for ASD Gene Prioritization
Background: This protocol describes the integration of ensemble methods with HGS optimization to enhance centrality measure validation in ASD gene discovery [51] [52].
Materials: Table: Essential Research Reagents and Computational Tools
| Item | Specification | Function/Purpose |
|---|---|---|
| Genomic Dataset | Whole genome/exome sequencing data from ASD cohorts [54] | Provides genetic variants for analysis |
| Protein Interaction Network | STRING database or InWeb_IM [43] [1] | Defines gene-gene interaction landscape |
| Brain Expression Data | BrainSpan Atlas [30] | Provides spatiotemporal expression context |
| HGS Framework | Multi-strategy HGS implementation [51] | Core optimization algorithm |
| Validation Gene Sets | SFARI Gene database [30] [54] | Gold standard for performance evaluation |
Procedure:
Data Preparation Phase (Duration: 2-3 days)
- Obtain genomic data from ASD cohorts (minimum recommended sample: 500 trios) [54]
- Annotate variants and filter for rare (MAF < 0.1%), likely gene-disrupting mutations
- Integrate protein-protein interaction data from STRING (combined score > 0.7) [43]
- Download and preprocess BrainSpan developmental transcriptome data [30]
HGS Initialization (Duration: 1-2 hours)
Optimization Phase (Duration: 2-5 days, depending on dataset size)
Validation Phase (Duration: 3-5 days)
Troubleshooting Tips:
- If convergence is too rapid, increase population size or chaos intensity parameters
- If biological validation fails, reweight fitness function to prioritize known ASD pathways
- For computational bottlenecks, implement binary HGS for feature selection [52]
Advanced Optimization Techniques
Hybrid similarity function architecture for improved gene prioritization
Implementation protocol for game theoretic centrality with HGS
Game theoretic centrality provides a novel approach to gene prioritization by evaluating the combinatorial influence of gene groups [26] [1]. When combined with HGS optimization, it significantly enhances ASD gene discovery.
Materials:
- Coalitional Game Theory (CGT) implementation [1]
- Protein-protein interaction network with confidence scores
- HGS optimization framework with multi-strategy support [51]
Procedure:
Network Preparation (Duration: 1 day)
- Construct comprehensive gene-gene interaction network
- Assign edge weights based on combined evidence (co-expression + physical interactions)
- Filter network to include only connections with strong biological support
Shapley Value Calculation (Duration: 1-2 days)
- Implement game theoretic centrality using Shapley value formulation [1]
- Compute marginal contribution of each gene to network connectivity
- Generate initial gene rankings based on synergistic influence
HGS Integration (Duration: 2-3 days)
Validation (Duration: 2 days)
Expected Results: This hybrid approach identifies influential genes that might be missed by conventional methods, particularly those in protein complexes and pathway bottlenecks relevant to ASD pathophysiology.
FAQ: Addressing Common Researcher Questions
How do I determine optimal HGS parameters for my specific ASD dataset?
Optimal parameters vary by dataset size and genetic architecture. Use this structured approach:
Start with established defaults [52]:
- Population size: 50-100
- Hunger sensitivity: α=50, β=0.5
- Maximum iterations: 500-1000
Perform sensitivity analysis:
- Test parameter ranges using a subset of your data
- Evaluate solution quality using convergence speed and biological validation
- Prioritize parameters that maximize replication in independent ASD cohorts
Implement adaptive parameters [51]:
- Use self-adjusting coefficients in oppositional learning
- Dynamically balance exploration/exploitation based on fitness improvement rates
What are the most effective ensemble strategies for HGS in ASD research?
Based on comparative studies [51] [52], the most effective ensemble strategies include:
Table: Effective Ensemble Strategies for HGS in ASD Gene Discovery
| Strategy | Mechanism | Advantage | Implementation Tip |
|---|---|---|---|
| Chaotic Initialization | Replaces random values with chaotic sequences | Improves population diversity by 25% [52] | Use Logistic map for initial population generation |
| Vertical Crossover | Exchanges gene segments between parents | Enhances exploitation without premature convergence [52] | Implement every 10 generations with elite preservation |
| Dynamic Oppositional Learning | Generates opposite solutions to escape local optima | Improves novel gene discovery by 18% [51] | Apply self-adjusting coefficients based on convergence stage |
| Adaptive Boundary Handling | Redirects out-of-bounds individuals to promising regions | Increases search efficiency by 22% [51] | Use quadratic interpolation for boundary redirection |
How can I handle missing or heterogeneous data in ASD genomic datasets?
Missing data is common in ASD genomics. Effective approaches include:
Federated learning approaches: Train models across multiple institutions without sharing raw data [53]
Transfer learning: Leverage pre-trained models from related neurodevelopmental disorders [53]
Multi-modal imputation: Integrate genetic, expression, and epigenetic data for informed missing value estimation [30]
HGS with uncertainty incorporation: Modify fitness functions to account for data reliability and completeness
What validation frameworks are most convincing for ASD gene discovery?
A tiered validation framework is recommended:
Computational validation:
Biological validation:
Clinical validation:
- Assess gene burden in large ASD cohorts (e.g., SPARK with 21,532 individuals) [54]
- Evaluate phenotypic specificity across neurodevelopmental conditions
Differentiating ASD-Specific Signals from General Neurodevelopmental Disruption
Analytical Framework and Core Concepts
Defining the Analytical Challenge
A primary challenge in autism spectrum disorder (ASD) research involves distinguishing molecular signals specific to ASD from those associated with general neurodevelopmental disruption. ASD is a complex neurodevelopmental disorder characterized by deficits in social communication and interaction, along with restricted interests and repetitive behaviors [55] [56]. The disorder has strong genetic underpinnings, with heritability estimated at approximately 50% and even higher in identical twins [57]. However, the genetic architecture is exceptionally heterogeneous, involving hundreds of risk genes and complex gene-environment interactions [57] [55] [30].
The clinical heterogeneity of ASD is reflected in its biological complexity, with multiple signaling pathways, neural circuits, and molecular mechanisms implicated in its pathogenesis [55] [58]. This complexity creates a significant methodological challenge: determining whether observed neurobiological alterations represent core ASD-specific pathology or secondary consequences of general neurodevelopmental disruption. This distinction is crucial for identifying valid therapeutic targets and developing effective interventions.
Key Signaling Pathways in ASD Pathogenesis
Table 1: Major Signaling Pathways Implicated in ASD
| Pathway | Key Components | Primary Functions | ASD-Specific Evidence |
|---|---|---|---|
| WNT/β-catenin | WNT1, WNT2, CTNNB1, APC, TCF7L2 | Neural patterning, synaptogenesis, axon guidance | Rare missense variants in WNT1 with enhanced signaling; APC mutations with autistic-like behaviors in mice [58] |
| BMP/TGF-β | BMPs, TGF-β receptors, SMADs | Neural differentiation, dendritic morphology | Interactions with ASD-associated genes (NLGN, UBE3A); dysregulated in some models [58] |
| SHH | SHH, PTCH1, SMO, GLI | Neural patterning, progenitor proliferation | Dysregulation linked to ASD pathogenesis; environmental factors affect pathway [58] |
| mTOR | TSC1/2, PTEN, FMRP | Protein synthesis, synaptic plasticity | Enlarged brains, hyperactive mTOR signaling in TSC1/2 models [57] |
| Metabotropic Glutamate | mGluR1/5, FMRP, GRM genes | Synaptic plasticity, protein translation | Targeted by investigational therapies for fragile X syndrome [55] |
Troubleshooting Guide: Frequently Encountered Experimental Challenges
FAQ: Establishing ASD-Specificity of Genetic Signals
Q: How can I determine whether a genetic signal is ASD-specific rather than general neurodevelopmental disruption?
A: Implement a multi-tiered analytical approach:
- Constraint metrics analysis: Calculate gene-level constraint metrics including pLI (probability of being loss-of-function intolerant), Z-scores for synonymous (synz), missense (misz), and LoF variants (lof_z) using large population databases like gnomAD [30]. ASD risk genes show significant intolerance to protein-truncating variants.
- Spatiotemporal co-expression analysis: Analyze whether candidate genes show enriched co-expression in specific brain regions (prefrontal cortex, striatum) during critical developmental windows (mid-fetal period, early childhood) using resources like BrainSpan Atlas [30]. True ASD genes often cluster in specific spatiotemporal patterns.
- Network centrality measures: Construct protein-protein interaction networks and calculate network centrality parameters (degree centrality, betweenness centrality) to identify hub-bottleneck genes [43]. True ASD genes often occupy central positions in biological networks.
- Cross-disorder comparison: Compare your candidate genes with gene sets from other neurodevelopmental disorders (intellectual disability, epilepsy) to identify ASD-specific signatures [30].
Q: What controls should I include when validating ASD-specific signaling pathway alterations?
A: Implement these essential experimental controls:
- Disease specificity controls: Compare pathway activity in ASD models versus models of other neurodevelopmental disorders with overlapping genetic etiology (e.g., compare SHANK3 mutants with models of general intellectual disability).
- Developmental stage controls: Analyze pathway activity across multiple developmental timepoints, as ASD-specific alterations often manifest during critical windows like mid-fetal development or early synaptogenesis [30] [58].
- Circuit-specific controls: Examine pathway function in neural circuits specifically implicated in ASD core symptoms (prefrontal-striatal, cerebellar-thalamic) versus control circuits [57] [55].
- Rescue experiment controls: Demonstrate that pathway-specific interventions rescue ASD-relevant behavioral phenotypes but do not affect general neurodevelopmental parameters.
FAQ: Technical Challenges in Pathway Analysis
Q: My signaling pathway experiments show inconsistent results across different ASD models. How can I resolve this?
A: Inconsistencies often arise from model system limitations and pathway crosstalk:
- Model selection strategy: Employ multiple, well-validated ASD models including genetic (SHANK3, NLGN, CHD8), environmental (VPA, maternal immune activation), and idiopathic (BTBR) models to distinguish consistent pathway alterations from model-specific effects [57].
- Pathway crosstalk accounting: Implement simultaneous monitoring of multiple interacting pathways (WNT, BMP, SHH), as crosstalk between pathways is common in ASD [58]. For example, UBE3A influences both WNT and BMP signaling.
- Cell-type specific resolution: Utilize cell-type specific profiling techniques (TRAP, snRNA-seq) to resolve pathway activity in specific neuronal populations, as bulk tissue analysis may mask cell-type specific effects [57] [55].
- Experimental rigor: Ensure consistent experimental conditions, especially developmental timepoints and brain regions, as pathway activity is highly spatiotemporally regulated.
Experimental Protocols for Signal Differentiation
Protocol: Differentiating ASD-Specific Transcriptional Signatures
Objective: Distinguish ASD-specific transcriptional patterns from general neurodevelopmental disruption signatures.
Methodology:
- Data Collection: Obtain transcriptomic data from:
- ASD genetic models (minimum 3 different high-confidence ASD genes)
- General neurodevelopmental disruption models (hypoxia, general synaptic dysfunction)
- Typically developing controls Across multiple developmental timepoints (E15, P7, P30, adult) [30]
Feature Selection:
- Calculate fold changes versus controls for all groups
- Identify genes differentially expressed in ASD models but unchanged in general disruption models
- Apply gene-level constraint metrics (pLI > 0.9) to prioritize constrained genes
Network Analysis:
- Construct co-expression networks using WGCNA or similar approaches
- Calculate network centrality measures (degree, betweenness) for all genes
- Identify hub genes unique to ASD networks [43]
Validation:
- Cross-reference with human ASD postmortem transcriptomic data
- Validate top candidates using spatial transcriptomics in relevant brain regions
- Test functional relevance using CRISPR-based functional assays
Table 2: Key Analytical Metrics for Differentiating ASD-Specific Signals
| Metric Category | Specific Measures | ASD-Specific Pattern | General Disruption Pattern |
|---|---|---|---|
| Genetic Constraint | pLI, LOEUF, missense Z-score | High constraint (pLI > 0.9) | Variable constraint |
| Spatiotemporal Expression | BrainSpan enrichment, developmental trajectories | Mid-fetal cortical enrichment, specific developmental patterns | Diffuse patterns, inconsistent timing |
| Network Properties | Degree centrality, betweenness centrality | High centrality in protein interaction networks | Peripheral network positions |
| Cross-Disorder Specificity | Odds ratios for ASD vs other NDDs | High ASD specificity | Shared across multiple disorders |
| Pathway Convergence | Enrichment in synaptic, chromatin, WNT pathways | Specific pathway convergence | Diffuse pathway involvement |
Protocol: Validating ASD-Specific Signaling Pathway Alterations
Objective: Confirm that observed signaling pathway alterations are specific to ASD pathophysiology rather than general neurodevelopmental disruption.
Methodology:
- Multi-Model Screening:
- Test pathway activity in a panel of ASD models (genetic, environmental, idiopathic)
- Include control models of general neurodevelopmental disruption
- Assess multiple pathway readouts (phosphorylation, target gene expression, localization)
Developmental Profiling:
- Map pathway activity across developmental trajectory (embryonic to adult)
- Identify critical windows of pathway dysfunction
- Correlate with behavioral development
Circuit-Specific Analysis:
- Use circuit-specific labeling techniques (virally-mediated Cre delivery)
- Assess pathway function in ASD-relevant circuits (cortico-striatal, social behavior network)
- Compare with control circuits
Intervention Studies:
- Implement pathway-specific pharmacological or genetic interventions
- Assess rescue of ASD-relevant behaviors
- Determine effects on general neurodevelopmental parameters
Key Signaling Pathways: Mechanisms and Crosstalk
The complexity of distinguishing ASD-specific signals arises from extensive crosstalk between major signaling pathways. Several key pathways demonstrate particularly important interactions in ASD pathogenesis:
WNT Signaling: Both canonical (β-catenin-dependent) and non-canonical WNT signaling are implicated in ASD, with evidence from both human genetics and animal models [58]. Key ASD-risk genes like CHD8 regulate WNT signaling, and β-catenin conditional knockouts show ASD-relevant behavioral phenotypes. WNT signaling demonstrates significant crosstalk with other pathways including BMP and RA signaling.
BMP/TGF-β Signaling: BMP signaling modulates neuronal differentiation and connectivity, with several ASD-associated genes (NLGN, UBE3A, FMR1) influencing BMP pathway activity [58]. The balance between BMP and WNT signaling appears particularly important for cortical development.
mTOR Pathway: The mTOR pathway integrates numerous ASD-relevant signals, with several monogenic ASD forms (TSC, PTEN, FMR1) directly affecting mTOR signaling [57] [55]. mTOR inhibitors represent one of the most promising targeted therapeutic approaches for specific ASD forms.
ASD Gene Network and Pathway Relationships
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Differentiating ASD-Specific Signals
| Reagent Category | Specific Examples | Application | Key Considerations |
|---|---|---|---|
| Genetic Models | SHANK3 KO, CHD8 KO, NLGN3 R451C, FMR1 KO, VPA exposure | Pathway analysis across etiologies | Include both monogenic and idiopathic models; control for background strain effects [57] |
| Cell Type Markers | PV, SST, VIP, CamKIIa, GFAP cre lines | Cell-type specific pathway analysis | Use multiple complementary markers; validate specificity [57] [55] |
| Pathway Reporters | TCF/LEF-GFP, BMP-SMAD reporter, mTOR activity sensors | Live monitoring of pathway activity | Confirm reporter sensitivity and dynamic range; use multiple reporters per pathway [58] |
| Centrality Analysis Tools | Cytoscape with NetworkAnalyzer, igraph, custom Python/R scripts | Network-based gene prioritization | Use multiple centrality measures; validate with bootstrap resampling [43] [30] |
| Spatiotemporal Databases | BrainSpan Atlas, PsychENCODE, Human Brain Transcriptome | Developmental expression profiling | Account for batch effects; use consistent normalization methods [30] |
| Constraint Metrics | gnomAD pLI scores, LOEUF, missense constraint Z | Gene-level intolerance assessment | Use most recent database versions; consider population stratification [30] |
ASD-Specific Signal Validation Workflow
Proving the Pipeline: Validation Frameworks and Comparative Performance Analysis
Troubleshooting Guides & FAQs
Frequently Asked Questions
Q1: What are the primary sources of false positives in de novo mutation (DNM) calling, and how can I mitigate them? False positives in DNM calling predominantly arise from sequencing artifacts, mapping artifacts, and uneven sequence coverage [59]. To mitigate these:
- Utilize trio-based deep learning models: Tools like DeNovoCNN convert sequence read alignments into images for classification, significantly improving precision by learning to recognize these artifacts [59].
- Implement stringent post-calling filtration: Filter variants based on quality metrics such as GATK quality score (e.g., >300 for substitutions, >500 for indels), read coverage (e.g., ≥20× in the proband), and variant allele frequency (e.g., VAF >30%) [59].
- Employ robust validation techniques: Orthogonal validation using methods like Sanger sequencing or PacBio HiFi sequencing is recommended to confirm high-confidence DNMs [59].
Q2: How can I prevent train/test leakage when building a DNM benchmarking dataset? Train/test leakage, where information from the training data unfairly influences the test results, can be prevented by ensuring peptide or variant disjointedness between training and test sets [60].
- Method: After generating a set of high-confidence variants, perform an all-against-all comparison to identify any variant (considering all modified forms of a sequence as identical) that appears in more than one sample or species. Assign each duplicated variant to a single set (e.g., training) and eliminate it from all others [60].
- Rationale: This prevents models from memorizing features of specific variants and ensures a fair evaluation of their generalizability [60].
Q3: My analysis identifies novel candidate genes. How can I contextually validate their biological significance? Validation should integrate computational prioritization with experimental evidence.
- Computational Prioritization: Construct a gene-interaction network from the literature and use network centrality measures (e.g., degree, eigenvector, betweenness) to rank genes. The hypothesis is that genes central to a network of known ASD-related genes are more likely to be causally involved [61].
- Experimental Validation:
- In vivo models: Generate and characterize mouse models with a heterozygous deletion of the candidate gene. Key tests include interactive social behavior assays (e.g., three-chamber test) and communication analysis (e.g., ultrasonic vocalizations) to assess ASD-relevant phenotypes [62] [63].
- Gene Expression Analysis: Use single-cell RNA sequencing to identify the specific cell types in which your candidate ASD-related genes are enriched, providing insight into the neurobiological mechanisms [63].
Q4: Why is a cohort of proband-parent trios essential for confident DNM identification? Trio sequencing (proband and both unaffected parents) provides a direct genetic control. DNMs are defined as variants that are present in the proband but completely absent from both parents' genomes [59] [63]. This design allows for the straightforward segregation of de novo events from the vast number of inherited polymorphisms and shared sequencing errors.
Experimental Protocols & Methodologies
Protocol 1: Building a Gold Standard DNM Set from Whole-Exome Sequencing (WES) Trios
This protocol outlines the steps for creating a high-confidence dataset for benchmarking, based on established methods [59].
- Sample Preparation & Sequencing: Perform WES on DNA from child-parent trios using standard platforms (e.g., Illumina HiSeq) and exome enrichment kits (e.g., Agilent SureSelect) [59].
- Data Processing & Alignment: Process raw sequencing data and align reads to a reference genome (e.g., using BWA-MEM).
- Variant Calling: Perform multi-sample variant calling on the trio. This can be done with tools like GATK, Samtools, or specialized DNM callers like DeNovoGear [59].
- Initial DNM Selection: Select variants where the proband's genotype is heterozygous (0/1) and both parents are homozygous reference (0/0).
- Quality Filtering: Apply stringent filters to the initial call set. A representative filter set includes [59]:
- GATK quality score >300 for single-nucleotide variants (SNVs).
- GATK quality score >500 for insertions/deletions (indels).
- Read coverage ≥20× in the proband.
- Variant allele frequency (VAF) >30% in the proband.
- Curation & Final Dataset Creation: Manually inspect candidate DNMs using software like the Integrative Genomics Viewer (IGV) to remove obvious inherited variants or artifacts. Classify variants as DNM, inherited variant (IV), or unknown (UN), removing the UN category to create a final gold standard set [59].
Protocol 2: A Deep Learning Workflow for DNM Calling (DeNovoCNN)
This protocol details a deep convolutional neural network (CNN) approach for improved DNM detection [59].
- Image Generation:
- Input: For each candidate variant, extract the read pileup data for the trio members (child, father, mother) from the BAM/CRAM files across a 41-base window (20 bases upstream/downstream).
- Encoding: Encode the data as a 160 (height, max reads) x 164 (width) RGB image.
- Width: Each genomic position is represented by 4 pixels, forming a one-hot vector for bases A, C, T, G.
- Color Channels: Red for the child, Green for the father, Blue for the mother.
- Intensity: Pixel intensity (0-255) is scaled by mapping and base quality scores [59].
- Model Architecture & Training:
- Use a CNN architecture with nine 2D convolutional layers (96 filters, 3x3 kernels), batch normalization, and Squeeze-and-Excitation blocks.
- Train separate models for substitutions, insertions, and deletions due to their distinct patterns.
- Optimize hyperparameters (learning rate, batch size) using an algorithm like Hyperband [59].
- Classification: The model performs a binary classification task, assigning each input image to either a DNM or an inherited variant (IV) class [59].
Data Presentation
Table 1: Performance Comparison of DNM Calling Methods on Test Dataset
This table summarizes the benchmarking results of various DNM callers against a gold standard set, demonstrating the performance gains of a deep learning approach [59].
| Method | Recall (Sensitivity) | Precision | Key Characteristics |
|---|---|---|---|
| DeNovoCNN (CNN) | 96.74% | 96.55% | Treats DNM calling as an image classification problem; requires trio BAM/CRAM files [59]. |
| DeepTrio | Data not provided in search results | Data not provided in search results | A deep learning method for variant calling that performs multi-sample calling [59]. |
| GATK | Lower than DeNovoCNN | Lower than DeNovoCNN | Standard multi-sample variant calling pipeline; DNMs are selected post-hoc based on genotypes [59]. |
| DeNovoGear | Lower than DeNovoCNN | Lower than DeNovoCNN | Uses a statistical model with mutation rate priors; can work from existing VCFs [59]. |
| Samtools | Lower than DeNovoCNN | Lower than DeNovoCNN | A traditional variant caller; in-house methods often use it as a base for custom filters [59]. |
Table 2: Essential Research Reagent Solutions for ASD Gene Discovery
A list of key materials and resources used in the cited studies for discovering and validating ASD genes [59] [62] [63].
| Research Reagent | Function in Research Context |
|---|---|
| Whole Exome Sequencing (WES) | Technique to identify coding variants, including de novo mutations, in proband-parent trios [62] [63]. |
| Trio WES Datasets | The foundational biological data (proband + both parents) required for confident DNM identification [59] [63]. |
| Sanger / PacBio HiFi Sequencing | Orthogonal validation technologies used to confirm the existence and zygosity of DNMs called from NGS data with high accuracy [59]. |
| Reference Proteomes (e.g., UniProt) | Curated protein sequence databases used as the target for database searches in mass spectrometry-based proteomics benchmarks [60]. |
| Mouse Models (e.g., Slc35g1+/-) | In vivo system for functional validation of candidate ASD genes through behavioral phenotyping (e.g., social interaction tests) [62] [63]. |
| Single-cell RNA Sequencing | Technology to profile gene expression in individual cell types, revealing where ASD-risk genes are co-expressed or enriched [63]. |
| Curation Software (e.g., IGV) | Allows for the visual inspection of sequence read alignments to manually curate and verify variant calls, forming a gold standard [59]. |
Workflow Visualizations
Gold Standard DNM Creation
Deep Learning DNM Calling
Candidate Gene Validation
This technical support center provides troubleshooting guides and experimental protocols for researchers validating computationally predicted Autism Spectrum Disorder (ASD) genes, particularly those identified through network centrality measures. The content addresses key challenges in linking candidate genes to specific ASD subtypes and underlying biological pathways, enabling more precise target discovery for therapeutic development.
Troubleshooting Guides and FAQs
FAQ: Subtype-Specific Genetic Validation
Q: Why is my candidate gene not showing clear phenotypic effects in animal models?
A: This commonly occurs when ASD heterogeneity is not accounted for in validation models. Recent research has identified four clinically and biologically distinct ASD subtypes with different genetic profiles [22]:
- Social and Behavioral Challenges subtype (37% of cases): Shows core autism traits without developmental delays, often with co-occurring conditions like ADHD and anxiety
- Mixed ASD with Developmental Delay subtype (19%): Features developmental milestone delays but fewer co-occurring psychiatric conditions
- Moderate Challenges subtype (34%): Exhibits milder core autism behaviors without co-occurring psychiatric conditions
- Broadly Affected subtype (10%): Presents with severe, wide-ranging challenges including developmental delays and multiple co-occurring conditions
Troubleshooting Recommendations:
- Match genetic profiles to subtypes: The Broadly Affected subtype shows the highest proportion of damaging de novo mutations, while only the Mixed ASD with Developmental Delay subtype carries more rare inherited variants [22]
- Consider developmental timing: Genes active in the Social and Behavioral Challenges subtype may become active later in childhood, unlike other subtypes where effects are predominantly prenatal [22]
- Use subtype-specific validation: Ensure your model system matches the developmental timeline and genetic profile of the target subtype
FAQ: Pathway Convergence Validation
Q: How can I determine if multiple candidate genes converge on common biological pathways?
A: This requires moving from single-gene to systems-level validation. Research reveals that ASD heterogeneity follows a "continuum moderated by subtype-common pathways" with distinctive profound autism driven by added subtype-specific embryonic pathways [64].
Troubleshooting Recommendations:
- Identify pathway gradients: Analyze whether pathway dysregulation follows severity gradients, with profound ASD showing greatest dysregulation and mild ASD showing least [64]
- Focus on embryonic pathways: For profound autism validation, prioritize pathways controlling proliferation, differentiation, neurogenesis, and DNA repair [64]
- Validate across modalities: Integrate clinical scores, gene expression, eye tracking, and fMRI data to confirm pathway-subtype relationships [64]
FAQ: Cross-Ancestry Validation Challenges
Q: Why do my candidate genes from European cohorts not replicate in other populations?
A: Genetic studies have predominantly focused on European and Hispanic ancestries, creating significant gaps in our understanding of ASD genetics across populations [62].
Troubleshooting Recommendations:
- Account for ancestral differences: Actively seek and incorporate data from diverse populations, such as the Chinese ASD cohort that identified novel candidate genes absent from current ASD databases [62]
- Validate functionally: Even without replication in different populations, use functional approaches like mouse models to confirm gene relevance, as demonstrated with SLC35G1 [62]
- Distinguish ASD with and without DD/ID: ASD without developmental delay or intellectual disability carries fewer disruptive de novo variants, affecting validation strategies [62]
Experimental Protocols for Biological Validation
Protocol 1: Subtype-Specific Gene Validation Workflow
Table 1: Key Experimental Approaches for Subtype-Specific Validation
| Method | Application | Key Parameters | Expected Outcomes |
|---|---|---|---|
| Whole-exome sequencing (1,141 trios) [62] | Identify novel candidate genes across ancestries | De novo variant analysis in large cohorts | 9+ novel ASD candidate genes beyond current databases |
| Single-cell RNA sequencing [62] | Identify cell types enriched for ASD-related genes | Cell type-specific expression patterns | Candidate gene expression in relevant neural populations |
| Mouse behavioral models [62] | Functional validation of social behavior effects | Heterozygous deletion, social interaction tests | Interactive social behavior defects (e.g., Slc35g1 models) |
| Similarity Network Fusion [64] | Integrate clinical and molecular data for subtyping | 12+ clinical and transcriptomic features | Identification of 4 ASD clusters with distinct molecular profiles |
Diagram 1: Subtype-Specific Gene Validation Workflow
Protocol 2: Pathway-Centric Validation Approach
Table 2: Pathway Analysis Methods for ASD Gene Validation
| Method | Purpose | Key Metrics | Interpretation Guidelines |
|---|---|---|---|
| MSigDB Hallmark pathway analysis [64] | Identify subtype-specific dysregulated pathways | 50 pathway activity scores from RNAseq | 7 embryonic pathways specific to profound autism |
| Protein-Protein Interaction networks [24] | Prioritize genes using systems biology | Betweenness centrality in PPI networks | Filter by brain expression for specificity |
| Multi-omics integration [64] | Link pathways to clinical outcomes | Social attention, fMRI, developmental trajectories | Pathway dysregulation severity correlates with clinical severity |
| Monte-Carlo validation [24] | Test statistical significance of network findings | p-value for SFARI gene enrichment (e.g., p < 2E-16) | Confirm non-random association with ASD |
Diagram 2: Pathway Relationships in ASD Subtypes
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Resources for ASD Gene Validation Studies
| Resource Type | Specific Examples | Application | Key Features |
|---|---|---|---|
| Genomic Databases | SFARI Gene Database [24] | Candidate gene prioritization | Curated ASD risk genes |
| Pathway Resources | MSigDB Hallmark Pathways [64] | Pathway enrichment analysis | 50 refined pathway signatures |
| Analysis Software | DataAssist/ExpressionSuite [65] | qPCR data normalization | Multiple endogenous control options |
| Cohort Resources | ABIDE I dataset [66] | Brain feature correlation | 419 structural MRI features |
| Experimental Models | Heterozygous mouse models [62] | Social behavior validation | Interactive social behavior tests |
| Sequencing Approaches | Whole-exome sequencing (1,141 trios) [62] | Novel gene discovery | Cross-ancestry validation |
| Network Tools | Protein-Protein Interaction networks [24] | Systems biology prioritization | Betweenness centrality analysis |
Advanced Technical Notes
Addressing Centrality Measure Limitations
When using betweenness centrality for gene prioritization [24]:
- Filter for brain specificity: Incorporate gene expression data from the Human Brain Tissue Bank to reduce network size while maintaining 94.3% of original nodes
- Validate statistically: Use Monte-Carlo approaches with 1,000 random seeds to confirm SFARI gene enrichment significance (p < 2E-16)
- Combine with functional data: Integrate spatiotemporal expression in ASD-relevant tissues and de novo mutation data
qPCR Validation Best Practices
For gene expression validation of candidate genes [67] [65]:
- Ensure PCR efficiency between 90-100% (-3.6 ≥ slope ≥ -3.3)
- Use multiple endogenous controls or global normalization when targets are numerous
- Implement automated liquid handling to reduce Ct value variations from manual pipetting
- Address non-specific amplification through primer redesign and annealing temperature optimization
FAQs and Troubleshooting Guides
Frequently Asked Questions
Q1: What is the primary purpose of a gene prioritization tool like forecASD in autism research?
Gene prioritization tools like forecASD are computational frameworks designed to analyze genomic data and identify which genetic variants are most likely to be pathogenic and contribute to Autism Spectrum Disorder (ASD). They help researchers sift through hundreds of potential candidate genes by integrating various lines of evidence, including the type of genetic variant, its population frequency, predicted functional impact, and whether it occurs de novo (newly formed in the affected individual) [23] [68]. Given that ASD may be associated with 400-1,000 genes, these tools are essential for managing this extreme genetic heterogeneity and focusing research on the most promising candidates [69] [70].
Q2: My analysis with a prioritization tool yielded a gene that is already a known, high-confidence ASD risk gene. Is this a valid result?
Yes, this is a valid and often expected result, especially when validating a tool's performance. A core part of validating a new prioritization method is to test it on established datasets and confirm that it can successfully identify known risk genes. Large-scale genomic studies have identified over 100 high-confidence ASD genes [23] [68]. Successfully flagging these genes demonstrates that your tool's algorithm and weighting criteria are functioning correctly and are aligned with biological reality.
Q3: What does a "low burden score" for a rare inherited variant mean, and how should I interpret it?
A low burden score indicates that a particular rare inherited variant is not statistically enriched in individuals with ASD compared to control populations. In the context of your analysis, it suggests that this specific variant may not be a major driver of the phenotype on its own [23]. However, interpretation requires caution. It could be a benign variant, or it could act in combination with other genetic factors (a polygenic contribution) or environmental influences to affect risk. You should not automatically dismiss a gene based on a single low-scoring variant, especially if it falls within a key biological pathway.
Q4: I've identified a potentially damaging variant in a non-coding region (e.g., an enhancer). Why didn't my prioritization tool rank it highly?
Many established prioritization tools, especially those built on earlier exome sequencing data, are primarily calibrated to assess the impact of variants within protein-coding genes [23] [68]. The interpretation of non-coding variants is more challenging because it requires additional data to predict their effect on gene regulation. Newer tools and whole-genome sequencing (WGS) studies are increasingly focusing on non-coding variants [23]. If your tool does not incorporate functional genomic data (like chromatin interaction maps or regulatory element annotations), it may undervalue these findings. For such variants, manual investigation and the use of specialized regulatory element prediction tools are recommended.
Q5: My cohort includes individuals of non-European ancestry. How might this affect the performance of my gene prioritization tool?
This is a critical consideration. Many existing genetic databases and the discovery cohorts for ASD genes are predominantly of European ancestry, which can introduce bias and reduce the accuracy of prioritization tools when applied to other populations [23] [70]. Tools may have higher false-negative rates in ancestrally diverse cohorts because they rely on frequency filters from biased reference databases. Your work in an ancestrally diverse cohort is a significant strength. To mitigate this, ensure you are using the most diverse population frequency databases available (like gnomAD) and be aware that you may discover novel, ancestry-specific risk variants that expand the genetic landscape of ASD [70].
Troubleshooting Common Experimental Issues
Issue 1: High Number of Candidate Genes After Initial Filtering
- Problem: The initial analysis pipeline has produced an unmanageably long list of candidate genes, making it difficult to select targets for downstream validation.
- Solution:
- Apply Functional Convergence Filters: A powerful strategy is to filter genes based on their involvement in shared biological pathways. Genes that are part of the same network (e.g., synaptic function, chromatin remodeling, or nervous system development) provide stronger evidence than a scattered list of genes [23] [68]. Use pathway enrichment tools (like DAVID, Enrichr) or protein-protein interaction networks (like STRING) to identify convergent biology.
- Integrate Expression Data: Filter your gene list against databases of brain-specific expression, such as BrainSpan, to prioritize genes that are active in relevant brain regions and during critical neurodevelopmental windows (e.g., the prenatal prefrontal cortex) [23] [70].
- Leverage Gene Constraint Metrics: Use metrics like pLI (probability of being loss-of-function intolerant) from gnomAD. Genes that are highly intolerant to protein-truncating variants are more likely to be involved in dominant disorders like ASD [23].
Issue 2: Discrepancy Between Tool Prediction and Functional Assay Results
- Problem: A variant ranked highly by the prioritization tool shows no observable effect in a preliminary cellular or animal model assay.
- Solution:
- Verify the Variant Call: Re-check the quality of the original sequencing data for that variant. Confirm the read depth and quality scores to rule out a sequencing artifact.
- Reassess the Biological Model: The functional assay may not fully capture the gene's role in human brain development. Consider if a different cell type (e.g., neuronal progenitors vs. mature neurons) or a more complex model (e.g., cerebral organoids) is needed.
- Investigate Genetic Context: The variant's effect might be modified by the individual's unique genetic background. The presence of protective or compensatory mechanisms in your model system could be masking the phenotype.
Issue 3: Handling Inconclusive or Conflicting Evidence for a Variant
- Problem: For a specific variant, some lines of evidence (e.g., de novo status) are strong, but others (e.g., population frequency, in silico prediction) are conflicting or of uncertain significance.
- Solution: Create a standardized evidence-weighted scoring system. The table below outlines how to quantify different types of evidence to help break ties between candidates.
Table: Evidence Weighting Scheme for Candidate Variant Interpretation
| Evidence Type | Strong Weight (e.g., +2) | Moderate Weight (e.g., +1) | Negative Weight (e.g., -1) |
|---|---|---|---|
| Inheritance | De novo in a sporadic case | Inherited from affected parent | Absent in affected family members (non-segregation) |
| Population Data | Absent from population controls (gnomAD) | Very low frequency (<0.001%) | Relatively common frequency (>0.01%) |
| Functional Prediction | Protein-truncating (PTV) in a constrained gene | Predicted damaging missense | Predicted benign |
| Previous Evidence | Known ASD gene [23] | Gene implicated in related NDD | No previous associations |
Experimental Protocols for Validation
Protocol 1: Validation of Gene Prioritization Output Using Established ASD Gene Sets
Objective: To benchmark the performance of the forecASD tool by determining its sensitivity and specificity in recovering known ASD risk genes.
Materials:
- Positive Control Set: A curated list of high-confidence ASD genes (e.g., from SFARI Gene database, categories 1 and 2).
- Negative Control Set: A list of genes not known to be associated with ASD or other neurodevelopmental disorders (NDDs), often sampled from genes tolerant to loss-of-function variation.
- Test Dataset: A whole-exome or whole-genome sequencing dataset from a well-characterized ASD cohort (e.g., SSC, SPARK) [23].
- Computational Resources: Workstation with forecASD installed and adequate processing power.
Methodology:
- Data Input: Run the positive control set, negative control set, and the test dataset through the forecASD pipeline using its standard parameters.
- Score Thresholding: Apply a score threshold to define "prioritized" genes from the forecASD output.
- Performance Calculation:
- Sensitivity (Recall): Calculate the proportion of known ASD genes in the positive control set that were correctly prioritized by forecASD.
Sensitivity = (True Positives) / (True Positives + False Negatives) - Specificity: Calculate the proportion of genes in the negative control set that were correctly excluded by forecASD.
Specificity = (True Negatives) / (True Negatives + False Positives) - Precision: Calculate the proportion of forecASD-prioritized genes in the positive control set that are true known ASD genes.
Precision = (True Positives) / (True Positives + False Positives)
- Sensitivity (Recall): Calculate the proportion of known ASD genes in the positive control set that were correctly prioritized by forecASD.
- Comparison: Repeat steps 1-3 for other competing prioritization tools to generate comparative performance metrics.
Protocol 2: Functional Convergence Analysis of Prioritized Gene Lists
Objective: To determine whether the genes prioritized by forecASD converge on specific biological pathways and cell types, adding functional validation to the computational predictions.
Materials:
- Gene List: The list of top-ranked candidate genes generated by forecASD from your analysis.
- Software: Functional enrichment tool (e.g., clusterProfiler, Enrichr), protein-protein interaction network analyzer (e.g., STRING).
- Data Resources: Brain transcriptomic data from BrainSpan Atlas, cell-type-specific expression data (e.g., from single-cell RNA-seq studies of human cortex) [23].
Methodology:
- Pathway Enrichment Analysis:
- Input the candidate gene list into an enrichment tool using a background of all genes expressed in the brain or all genes in the human genome.
- Identify Gene Ontology (GO) terms (e.g., "synaptic transmission," "chromatin organization") and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways that are statistically over-represented.
- Co-expression Network Analysis:
- Use data from the BrainSpan Atlas to test if the candidate genes are co-expressed in specific modules, particularly those active during mid-fetal development, a critical period for ASD [23].
- Cell-Type Enrichment:
- Map the candidate gene list onto single-cell RNA-seq datasets from the developing human brain. Test for enrichment in specific neuronal lineages (e.g., cortical projection neurons in deep layers 5/6) or glial cells [23].
The workflow for this protocol can be summarized as follows:
The Scientist's Toolkit: Research Reagent Solutions
Table: Essential Resources for ASD Gene Discovery and Validation
| Item / Resource | Function / Application | Example(s) / Notes |
|---|---|---|
| Whole Exome/Genome Sequencing Data | Foundation for discovering coding and non-coding variants associated with ASD. | MSSNG, SPARK, SSC cohorts [23]; Ancestrally diverse cohorts to reduce bias [70]. |
| Variant Annotation Databases | Provides population frequency and evolutionary constraint data for filtering variants. | gnomAD [69], ExAC, 1000 Genomes [70]. pLI score is critical for assessing gene intolerance. |
| In Silico Prediction Algorithms | Computationally predicts the functional impact of missense and non-coding variants. | SIFT, PolyPhen-2 [70]. Combine multiple algorithms for robustness. |
| ASD Gene Databases | Curated repositories of known and candidate ASD genes for benchmarking and validation. | SFARI Gene. Use high-confidence genes as a positive control set. |
| Functional Genomic Datasets | Provides data on gene expression and regulation in the brain across development. | BrainSpan Atlas (transcriptomics) [70], PsychENCODE (epigenomics). |
| Statistical Genetics Tools | Identifies genes with a significant burden of rare variants in case-control cohorts. | TADA (Transmission And De novo Association) framework [23]. |
| Pathway & Network Analysis Tools | Identifies functional convergence among candidate genes. | STRING (protein interactions), DAVID/Enrichr (pathway enrichment) [68]. |
Data Presentation: Comparative Metrics
Table: Comparative Framework for forecASD and Alternative Prioritization Approaches
| Feature / Metric | forecASD (Hypothesized) | TADA-based Methods | Pathway-Centric Tools | WGS-Native Tools |
|---|---|---|---|---|
| Core Methodology | Centrality measures in integrated biological networks. | Bayesian model of de novo and rare inherited variant burden [23]. | Enrichment in predefined biological pathways or co-expression modules. | Genome-wide variant calling including non-coding regions. |
| Primary Data Input | WES/WGS-derived variant lists, protein-protein interactions, expression data. | WES/WGS data from parent-child trios or case/control cohorts. | A list of candidate genes. | Whole-genome sequencing data. |
| Strengths | Captures functional convergence; potentially higher specificity for polygenic contributions. | Statistically robust for high-effect de novo variants; established discovery record [23] [68]. | Provides immediate biological insight and testable hypotheses. | Comprehensive; can detect non-coding variants, STRs, and complex structural variants [23]. |
| Limitations | Performance depends on the quality and completeness of underlying network data. | Less effective for inherited variants and polygenic risk; requires large sample sizes. | May miss novel genes outside known pathways; reliant on pathway definitions. | Computationally intensive; interpretation of non-coding variants remains challenging. |
| Ideal Use Case | Prioritizing genes from WES studies of modest size or for identifying pathway-level disruptions. | First-tier analysis in large trio cohorts (thousands) for novel gene discovery. | Functional interpretation of gene lists from primary analyses. | Discovery of novel variant types in well-powered cohorts where WES is negative. |
| Handling of Non-Coding Variants | Possible if regulatory networks are integrated. | Limited, as primarily designed for coding variation. | Not a primary focus. | A core strength, identifies variants in enhancers/promoters [23]. |
Frequently Asked Questions
Q1: What is the primary purpose of using cross-validation in ASD genomic studies? Cross-validation (CV) is a set of data sampling methods used to avoid overoptimism in overfitted models. Its primary purposes are to estimate an algorithm's generalization performance, select the best algorithm from several candidates, and tune model hyperparameters. By repeatedly partitioning a dataset into independent training and test cohorts, CV helps ensure that performance measurements are not biased by direct overfitting of the model to the data [71].
Q2: I have a limited dataset. Which CV method should I use to get the most reliable performance estimate? For smaller datasets, a repeated k-fold CV is highly recommended. While a simple k-fold CV (with k=5 or k=10) is standard, performing it multiple times with new random splits helps lower the variance of your estimate. The final performance score should be the average of all runs, leading to a more robust and reliable model selection [72].
Q3: What is a "data leak" and how can I prevent it during cross-validation? A data leak occurs when information from your test set is inadvertently used during the model training process. A common example is performing feature selection on the entire dataset before applying CV. This allows information about the test set to influence the model, leading to over-optimistic performance.
- Solution: Always integrate all steps, including feature selection and data normalization, inside the CV loop. Perform these operations solely on the training set of each split, then apply the selected features or fitted scaler to the test set [72] [73].
Q4: My model performs well in intra-cohort CV but fails on an independent cohort. What does this mean? This typically indicates that your model has learned patterns that are specific to the population from which your initial dataset was drawn. It may be picking up on technical artifacts or population-specific biological effects that do not generalize. When both intra-cohort and cross-cohort CV results are good, you can be more confident that your model has captured a more generalizable, biological signal [73].
Q5: How should I handle highly imbalanced classes in an ASD genomics dataset? Using standard k-fold CV on imbalanced data can result in splits that are not representative of the overall class distribution. The recommended solution is to use stratified k-fold CV, which preserves the percentage of samples for each class in every fold. However, note that this method does not account for other structures in your data, such as groups or families [72].
Q6: How do I apply cross-validation to family-based genetic data where individuals are not independent? Standard CV assumes that data points are independent. In family-based cohorts like SPARK or MSSNG, this assumption is violated. The solution is to use group k-fold CV, where the "group" is the family unit. This ensures that all samples from the same family are kept together in either the training or test set, preventing information leakage and providing a more realistic performance estimate [72].
Troubleshooting Guides
Problem 1: Overly Optimistic Performance During Validation
- Symptoms: Your model achieves high accuracy during cross-validation but performs poorly on a final, locked-away test set or an external cohort.
- Likely Causes:
- Tuning to the test set: Repeatedly modifying your model based on the CV test set performance causes the model to indirectly learn the noise and specifics of that test data [71].
- Data leakage: Information from the test set is leaking into the training process, often through improper preprocessing or feature selection conducted on the entire dataset before splitting [72].
- Non-representative test sets: The splits created during CV are not representative of the broader population due to small dataset size or hidden subclasses [71].
- Solutions:
- Use a separate holdout test set: Keep a test set completely separate from the CV process. Use it only for the final evaluation of your chosen model [72].
- Apply nested cross-validation: If you must use the entire dataset for both model selection and evaluation, use nested CV. An outer loop estimates generalization error, while an inner loop is dedicated to model selection and hyperparameter tuning, keeping the outer test data pristine [71].
- Audit your preprocessing pipeline: Ensure that all steps (e.g., normalization, feature selection) are fit solely on the training data of each CV fold and then applied to the test data [74].
Problem 2: High Variance in Cross-Validation Scores
- Symptoms: The performance metric (e.g., accuracy) varies widely across different folds of the CV.
- Likely Causes:
- Small dataset size: With limited data, a single fold can have a significant impact on the overall score.
- Improper number of folds (
k): With a highk(e.g., Leave-One-Out CV on a small dataset), each estimate has high variance [72].
- Solutions:
- Repeat the CV: Instead of running k-fold CV once, run it multiple times (e.g., 5 or 10 times) with different random seeds and average the results. This is known as repeated k-fold CV and provides a more stable estimate [72].
- Adjust the number of folds: Experiment with different values of
k. A common starting point is k=5 or k=10. Using a lower number of folds (e.g., k=5) can reduce variance, though it may slightly increase bias [71] [72].
Problem 3: Poor Generalization Across Diverse Populations
- Symptoms: A model trained and validated on one ancestral group (e.g., European) fails to perform on another (e.g., East Asian or African).
- Likely Causes:
- Ancestry-specific signals: The model has learned genetic variants or patterns that are specific to the population it was trained on, which may have different frequencies or be absent in other groups [75] [76].
- Lack of ancestral diversity in training data: Many large genomic datasets, including initial releases of MSSNG, SSC, and SPARK, have been predominantly of European ancestry [75] [23] [76].
- Solutions:
- Perform cross-cohort validation: Use one cohort (e.g., SPARK) for training and a different, ancestrally distinct cohort (e.g., the Korean autism cohort) for testing [73] [75].
- Utilize diverse datasets: Actively seek out and incorporate datasets with diverse ancestry, such as the Korean autism family cohort [75] or the Genomics of Autism in Latin American Ancestries Consortium [23], into your training or validation pipelines.
Major ASD Genomic Datasets for Cross-Validation
The table below summarizes key large-scale genomic datasets used in Autism Spectrum Disorder (ASD) research, which are pivotal for intra-cohort and cross-cohort validation studies.
| Dataset / Cohort Name | Primary Ancestry | Key Features & Data Types | Sample Size (Individuals) | Primary Use in CV |
|---|---|---|---|---|
| MSSNG [77] [23] | European | WGS data; includes SNVs, indels, SVs, tandem repeats | 11,312 | Intra-cohort validation; discovery of novel variants |
| SPARK [78] [23] | European | WGS & WES; extensive phenotypic data; large family-based cohort | >380,000 (registered); >40,000 (genotyped) [78] [23] | Large-scale training; internal & cross-cohort testing |
| Simons Simplex Collection (SSC) [23] | European | WGS & WES; simplex families | 9,205 [77] | Model development and tuning |
| Korean Autism Cohort [75] | East Asian | WGS; deep phenotyping; family-wise data | 2,255 (WGS); 3,730 (phenotypes) | Cross-cohort validation; testing generalizability |
| Genomics of Autism in Latin American Ancestries [23] | Admixed (Latin American) | WES & WGS | 15,427 | Enhancing ancestral diversity in training & testing |
Experimental Protocol: Cross-Cohort Validation
This protocol outlines the steps for performing a cross-cohort validation to test the generalizability of a genomic model for ASD.
Objective: To determine if a model trained on one population (e.g., of European ancestry) retains predictive performance on an independent population of different ancestry (e.g., East Asian).
Datasets:
Methodology:
- Feature Engineering: Perform feature selection (e.g., select top ASD-associated genes or variants) using only the training cohort (SPARK).
- Model Training: Train your chosen classifier (e.g., a linear SVM or Random Forest) on the entire processed training cohort.
- Model Application: Apply the fitted feature selector and trained model to the Korean Autism Cohort. Ensure that no information from the test cohort is used at this stage.
- Performance Evaluation: Calculate performance metrics (e.g., AUC, accuracy) on the Korean test set.
Interpretation:
- A significant drop in performance from the internal CV estimates (on SPARK) to the cross-cohort test (on the Korean cohort) suggests the model has poor generalizability and may be capturing population-specific signals.
- Similar performance indicates a more robust, generalizable biological signal has been learned [73].
Workflow Diagram: Cross-Cohort Validation
The following diagram illustrates the logical workflow for a cross-cohort validation study, which is essential for assessing the generalizability of findings in ASD genomics.
| Resource / Solution | Function in ASD Genomics Research |
|---|---|
| Whole-Genome Sequencing (WGS) | Enables comprehensive detection of coding and noncoding variants, including SNVs, indels, and structural variants [77] [23]. |
| Transmission and De Novo Association (TADA) | A Bayesian statistical framework for identifying genes with a significant burden of de novo and rare inherited mutations from sequencing data [23]. |
| Polygenic Score (PS) | Quantifies the cumulative contribution of common genetic variants to an individual's liability for a trait, used for risk stratification [75] [23]. |
| Stratified/Group K-Fold CV | A cross-validation method that preserves class distribution (stratified) and keeps correlated samples (e.g., from the same family) together in a single fold (grouped) [72]. |
| Ancestrally Diverse Cohorts | Datasets from non-European populations (e.g., Korean, Latin American) that are critical for testing and ensuring the generalizability of discovered genetic signals [75] [23]. |
Troubleshooting Guides and FAQs
Frequently Asked Questions
Q1: Our in silico predictions identified a novel candidate gene, but we are unsure how to begin functional validation. What is a typical workflow? A: A standard validation pipeline progresses from computational prediction to in vivo models. The case of SLC35G1 provides an excellent template [79] [63]:
- In Silico Prediction: The gene was first identified as a candidate through whole-exome sequencing of a large ASD cohort and bioinformatics analysis [63].
- In Vitro Characterization: Its function was clarified through uptake assays in transfected cell lines (e.g., HEK293, MDCKII), which defined its role as a citrate transporter [79] [80].
- In Vivo Validation: A mouse model with a heterozygous deletion of Slc35g1 was created, which exhibited defects in interactive social behaviors, confirming its relevance to ASD pathophysiology [63].
Q2: Our transport assay results for SLC35G1 are inconsistent. What could be a critical factor we are missing? A: SLC35G1 is highly sensitive to chloride ions. Its citrate transport activity is extensively inhibited by extracellular Cl− at physiologically relevant concentrations (IC50 = 6.7 mM) [79] [80]. Ensure your assay buffers carefully mimic the ionic conditions of your target biological environment (e.g., cytosolic vs. extracellular). For basolateral transport studies, the presence of extracellular Cl- (~120 mM) suggests SLC35G1 functions as a citrate exporter under physiological conditions [79].
Q3: How can we determine if a candidate gene is specifically associated with ASD, rather than general neurodevelopmental delay? A: Focus on patient cohorts and model systems that separate these features. The discovery of SLC35G1 as an ASD risk gene was strengthened by analyzing probands with and without developmental delay (DD)/intellectual disability (ID). Genes identified in cohorts without DD/ID may be more specific to core social dysfunctions [63]. In model organisms, test behavioral phenotypes beyond cognitive tasks, such as the social interaction deficits observed in Slc35g1 heterozygous mice [63].
Troubleshooting Common Experimental Issues
| Problem | Possible Cause | Suggested Solution |
|---|---|---|
| Low signal-to-noise ratio in [14C]citrate uptake assays [79] | Non-specific background transport or suboptimal expression. | Use a cell line with low endogenous transporter activity (e.g., HEK293). Establish stable transfectants to ensure consistent expression. Perform assays in Cl--free buffer to maximize specific uptake signal [79]. |
| Discrepancy between subcellular localization in your study vs. literature [79] [80] | Tagging protein may alter trafficking (e.g., GFP-tagged SLC35G1 was wrongly directed to ER). | Use immunofluorescence with validated antibodies against the endogenous protein. Test different tag locations (N- vs. C-terminal) or use untagged proteins for localization studies [80]. |
| Polarized cell model (e.g., MDCKII) shows no functional transport [79] | Transporter may be mis-sorted or not correctly integrated into the target membrane (apical vs. basolateral). | Verify the polarization and membrane integrity of your cell monolayer. Use immunohistochemistry with markers for apical and basolateral membranes (e.g., ATP1A1 for basolateral) to confirm correct SLC35G1 localization [79]. |
| Mouse model does not recapitulate expected ASD-like behaviors | Incomplete penetrance, compensatory mechanisms, or species-specific differences. | Consider generating heterozygous models, as a heterozygous deletion of Slc35g1 was sufficient to produce social defects in mice [63]. Employ a battery of behavioral tests to assess different ASD core features. |
The functional characterization of SLC35G1 yielded key kinetic and inhibitory parameters, summarized below.
Table 1: Key Functional Parameters of SLC35G1
| Parameter | Value | Experimental Context | Source |
|---|---|---|---|
| Km (for Citrate) | 519 μM | Uptake in MDCKII cells, pH 5.5, Cl--free buffer | [79] [80] |
| Vmax | 1.10 nmol/min/mg protein | Uptake in MDCKII cells, pH 5.5, Cl--free buffer | [79] [80] |
| IC50 (for Cl−) | 6.7 mM | Inhibition of citrate uptake in transfected cells | [79] [80] |
| pH Dependence | Uptake increased at acidic pH | Uptake was higher at pH 5.5 vs. pH 7.4 | [79] |
| Membrane Potential | Independent | Replacing Na+ with K+ had no impact on uptake | [79] |
Table 2: SLC35G1 Expression Profile and Genetic Association
| Aspect | Finding | Significance | Source |
|---|---|---|---|
| Tissue Expression | Highest in duodenum and jejunum; also in testis, pancreas | Supports primary role in intestinal citrate absorption | [79] [80] |
| Cellular Localization | Basolateral membrane of enterocytes and polarized Caco-2 cells | Identifies its role in citrate efflux into bloodstream | [79] |
| Genetic Association | Novel ASD candidate gene from Chinese trio WES study | Links gene to neurodevelopmental disorder | [63] [23] |
| In Vivo Validation | Social behavior defects in Slc35g1 heterozygous mice | Confirms role in behavior relevant to ASD pathology | [63] |
Experimental Protocols
Detailed Methodology: SLC35G1 [14C]Citrate Uptake Assay in Transfected Cells
This protocol is adapted from studies that characterized SLC35G1-mediated citrate transport [79] [80].
1. Cell Culture and Transfection:
- Culture model cells such as HEK293 or MDCKII in standard media.
- Transiently or stably transfect with a plasmid vector containing the full-length human SLC35G1 cDNA. Include a mock transfection (empty vector) control in every experiment.
- For polarized transport studies, culture MDCKII cells on Transwell permeable filters until fully polarized.
2. Uptake Assay Buffer Preparation:
- Uptake Buffer (Cl--free): To measure maximum transport activity, use a Hanks'-based solution with Cl- salts replaced by gluconate salts (e.g., Na-gluconate, K-gluconate). Adjust pH to 5.5 to enhance uptake signal for kinetic analysis.
- Inhibition Buffer (Cl--containing): Use standard Hanks' balanced salt solution (containing ~144 mM Cl-) to test chloride sensitivity.
3. Uptake Measurement:
- Wash cells twice with pre-warmed assay buffer.
- Incubate cells at 37°C with uptake buffer containing radiolabeled [14C]citrate. For kinetic studies, use a range of cold citrate concentrations spiked with a fixed amount of [14C]citrate.
- Terminate the uptake reaction at designated time points (e.g., 10 minutes) by rapidly washing cells with ice-cold buffer.
- Lyse cells and measure the accumulated radioactivity using a liquid scintillation counter. Normalize counts to total protein concentration.
4. Data Analysis:
- Calculate SLC35G1-specific uptake by subtracting uptake in mock-transfected cells from uptake in SLC35G1-expressing cells.
- For kinetic parameters (Km, Vmax), fit the specific uptake data to the Michaelis-Menten equation using non-linear regression.
Detailed Methodology: Functional Validation in Mouse Models
This protocol outlines the key steps for validating ASD candidate genes in vivo, as demonstrated for Slc35g1 [63].
1. Animal Model Generation:
- Generate a heterozygous knockout mouse model using CRISPR/Cas9 or homologous recombination to mimic the haploinsufficiency often observed in ASD. Confirm the genotype by PCR and sequencing.
2. Behavioral Phenotyping: Subject age-matched wild-type and heterozygous mice to a battery of behavioral tests, with a focus on ASD-relevant phenotypes:
- Social Interaction Test: Assess direct social interaction between two mice in a novel environment. A significant reduction in sniffing, following, and other social contacts in mutant mice indicates social deficits.
- Three-Chamber Sociability Test: Evaluate the mouse's preference for a novel mouse over a novel object. Mice with ASD-related phenotypes typically show no preference for the social stimulus.
- Repetitive Behavior Analysis: Quantify marble burying or self-grooming behavior, as an increase in these can indicate repetitive/stereotyped behavior.
- Control Tests: Perform additional tests like open field (for general activity and anxiety) and rotarod (for motor coordination) to ensure behavioral changes are specific to social and repetitive domains.
3. Analysis and Interpretation:
- Analyze data using appropriate statistical tests (e.g., t-test, ANOVA) comparing heterozygous mutants to wild-type controls.
- A specific deficit in social interaction without general motor or anxiety abnormalities provides strong evidence for a role of the gene in ASD-related pathways.
Visualization of Workflows and Pathways
SLC35G1 Validation Workflow
Citrate Transport Pathway in Gut
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for Transporter Validation
| Reagent / Material | Function / Application | Example from SLC35G1 Studies |
|---|---|---|
| Heterologous Expression Systems | Provides a controlled environment to study gene function in isolation. | HEK293 cells for initial functional screening; MDCKII cells for polarization and transwell transport studies [79] [80]. |
| Polarized Cell Culture Models | Enables study of transporter localization and directional solute transport. | MDCKII or Caco-2 cells cultured on Transwell filters to model apical and basolateral membranes [79]. |
| Radiolabeled Substrates | Allows sensitive and quantitative measurement of transporter activity. | [14C]Citrate used in uptake assays to directly measure SLC35G1 transport kinetics [79] [80]. |
| Ion-Specific Assay Buffers | Used to determine ion dependence and driving forces of transport. | Cl--free buffers to unmask SLC35G1 activity; Na+-free or K+-rich buffers to test membrane potential dependence [79]. |
| Validated Antibodies | Critical for determining protein localization and expression levels. | Antibodies against SLC35G1 used for immunohistochemistry to confirm basolateral localization in human jejunum [79]. |
| Genetically Engineered Mouse Models | The gold standard for in vivo functional validation of candidate genes. | Slc35g1 heterozygous knockout mice used to confirm its role in social behavior [63]. |
Conclusion
The validation of centrality measures marks a paradigm shift in ASD gene discovery, moving from isolated gene lists to a systems-level understanding of disrupted biological networks. The synthesis of foundational principles, robust methodologies, and rigorous validation, as demonstrated by tools like forecASD and the subtyping from recent studies, provides a powerful, multi-dimensional framework. Future directions must focus on expanding diverse ancestral representation in datasets, integrating non-coding genomic regions, and translating these validated genetic insights into biologically relevant subtypes for precision medicine. This approach holds the promise of uncovering the complex etiologies of ASD, ultimately guiding the development of targeted therapies and personalized diagnostic tools.
References
- 1. Game theoretic : a novel approach to prioritize disease... centrality [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-020-03693-1]
- 2. A systematic survey of centrality measures for protein-protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6069823/]
- 3. Revisiting the use of graph centrality models in biological ... [https://biodatamining.biomedcentral.com/articles/10.1186/s13040-020-00214-x]
- 4. Peripheral gene interactions define interpretable clusters of core ASD ... [https://www.nature.com/articles/s41540-022-00240-x?error=cookies_not_supported]
- 5. Advances in Genetic Discovery and Implications for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6192539/]
- 6. Recent Genetic and Functional Insights in Autism Spectrum ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6959126/]
- 7. Genetic Causes and Modifiers of Autism Spectrum Disorder [https://www.frontiersin.org/journals/cellular-neuroscience/articles/10.3389/fncel.2019.00385/full]
- 8. New Study Reveals Subclasses of Autism by Linking Traits ... [https://www.simonsfoundation.org/2025/07/09/new-study-reveals-subclasses-of-autism-by-linking-traits-to-genetics/]
- 9. Hybrid deep learning method to identify key genes in autism ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12023764/]
- 10. Polygenic and developmental profiles of autism differ by ... [https://www.nature.com/articles/s41586-025-09542-6]
- 11. Centrality [https://en.wikipedia.org/wiki/Centrality]
- 12. Evolution of Centrality Measurements for the Detection ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4999434/]
- 13. A Machine Learning Approach to Predicting Autism Risk Genes [https://pmc.ncbi.nlm.nih.gov/articles/PMC7513695/]
- 14. Network Centrality: Understanding Degree, Closeness & ... [https://visiblenetworklabs.com/2021/04/16/understanding-network-centrality/]
- 15. Centrality analysis in a drug network and its application to ... [https://www.sciencedirect.com/science/article/abs/pii/S0096300320308237]
- 16. 6 Vertex Importance and Centrality [https://ona-book.org/vertex-importance.html]
- 17. Centrality Analysis Methods for Biological Networks and Their ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2733090/]
- 18. Network analysis of protein interaction data [https://www.ebi.ac.uk/training/online/courses/network-analysis-of-protein-interaction-data-an-introduction/building-and-analysing-ppins/topological-ppin-analysis/centrality-analysis/]
- 19. A Mini Review of Node Centrality Metrics in Biological ... [https://www.sciltp.com/journals/ijndi/articles/2504000009]
- 20. Centrality-based pathway enrichment: a systematic approach ... [https://bmcsystbiol.biomedcentral.com/articles/10.1186/1752-0509-6-56]
- 21. Decomposition of phenotypic heterogeneity in autism ... [https://www.nature.com/articles/s41588-025-02224-z]
- 22. Major autism study uncovers biologically distinct subtypes, ... [https://www.princeton.edu/news/2025/07/09/major-autism-study-uncovers-biologically-distinct-subtypes-paving-way-precision]
- 23. Advancing precision diagnosis in autism: Insights from large ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12275934/]
- 24. A Systems Biology Approach for Prioritizing ASD Genes in ... [https://www.mdpi.com/1422-0067/26/5/2078/review_report]
- 25. A Systems Biology Approach for Prioritizing ASD Genes in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11900372/]
- 26. Game theoretic centrality: a novel approach to prioritize ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7430867/]
- 27. Graph Node Classification to Predict Autism Risk in Genes [https://pmc.ncbi.nlm.nih.gov/articles/PMC11049455/]
- 28. SFARI Gene - Welcome [https://gene.sfari.org/]
- 29. Multimodal Morphometric Similarity Network Analysis of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11940302/]
- 30. A Machine Learning Approach to Predicting Autism Risk ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2020.500064/full]
- 31. A hybrid Stacking-SMOTE model for optimizing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10559615/]
- 32. -wide prediction of dominant and recessive... Genome [https://pubmed.ncbi.nlm.nih.gov/40015282/]
- 33. a web server for brain expression Spatio-temporal pattern ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6896511/]
- 34. Spatial dynamics of brain development and ... [https://www.nature.com/articles/s41586-025-09663-y]
- 35. Spatiotemporal transcriptome atlas reveals the regional ... [https://www.sciencedirect.com/science/article/pii/S0092867423012333]
- 36. Cleaning, Analysis and Visualization of Graph Neural ... [https://medium.com/h7w/introduction-to-network-analysis-in-python-cleaning-analysis-and-visualization-of-graph-networks-c360e51274d4]
- 37. Social Network Analysis: Understanding Centrality Measures [https://cambridge-intelligence.com/keylines-faqs-social-network-analysis/]
- 38. FAQ - STRING Help [https://string-db.org/help/faq/]
- 39. How are the scores computed? [https://version10.string-db.org/help/faq/]
- 40. STRING 7—recent developments in the integration and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1669762/]
- 41. Node centrality measures are a poor substitute for causal ... [https://www.nature.com/articles/s41598-019-43033-9]
- 42. Genome-wide association studies in ancestrally diverse ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6939869/]
- 43. Identification of the Key Genes of Autism Spectrum Disorder ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8343959/]
- 44. SPARK [https://www.sfari.org/resource/spark/]
- 45. [PDF] SMOTE-RUS : Combined Oversampling and ... [https://www.semanticscholar.org/paper/SMOTE-RUS-%3A-Combined-Oversampling-and-Undersampling-Ismail-Gad/8b5180e060cef5d37d1fe6afdfcbe703e6904528]
- 46. Equitable machine learning counteracts ancestral bias in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11894161/]
- 47. Improving genetic risk prediction across diverse population ... [https://www.nature.com/articles/s42003-023-05352-6]
- 48. 9 Network Centrality and Hierarchy | Network Analysis [https://inarwhal.github.io/NetworkAnalysisR-book/ch9-Network-Centrality-Hierarchy-R.html]
- 49. The Constrained Disorder Principle - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC12650003/]
- 50. Challenges and opportunities in the network medicine of ... [https://www.sciencedirect.com/science/article/pii/S2666634025003472]
- 51. A multistrategy improved hunger games search algorithm [https://www.nature.com/articles/s41598-025-16513-4]
- 52. Multi-strategy ensemble binary hunger games search for ... [https://www.sciencedirect.com/science/article/abs/pii/S0950705122003707]
- 53. Applications of machine learning in drug discovery and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6552674/]
- 54. Return of genetic research results in 21532 individuals with ... [https://www.sciencedirect.com/science/article/pii/S1098360024001369]
- 55. Signalling pathways in autism spectrum disorder [https://www.nature.com/articles/s41392-022-01081-0]
- 56. Autism Spectrum Disorder - National Institute of Mental Health [https://www.nimh.nih.gov/health/publications/autism-spectrum-disorder]
- 57. Autism Spectrum Disorder: Neurodevelopmental Risk Factors ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9915249/]
- 58. Impaired neurodevelopmental pathways in autism spectrum ... [https://jneurodevdisorders.biomedcentral.com/articles/10.1186/s11689-019-9268-y]
- 59. a deep learning approach to de novo variant calling in next ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9508836/]
- 60. A multi-species benchmark for training and validating mass ... [https://www.nature.com/articles/s41597-024-04068-4]
- 61. Identifying gene-disease associations using centrality on a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2718658/]
- 62. Discovery and Validation of Novel Genes in a Large ... [https://pubmed.ncbi.nlm.nih.gov/37393044/]
- 63. Discovery and Validation of Novel Genes in a Large ... [https://www.sciencedirect.com/science/article/abs/pii/S0006322323013987]
- 64. Subtype-Specific Molecular Insights into Autism Spectrum ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11469458/]
- 65. Gene Expression Support—Troubleshooting [https://www.thermofisher.com/us/en/home/technical-resources/technical-reference-library/real-time-digital-PCR-applications-support-center/gene-expression-support/gene-expression-support-troubleshooting.html]
- 66. Node Centrality Measures Identify Relevant Structural MRI ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8071038/]
- 67. qPCR Troubleshooting: How to Ensure Successful ... [https://dispendix.com/blog/qpcr-troubleshooting]
- 68. Genetic architecture of autism spectrum disorder [https://www.sciencedirect.com/science/article/pii/S0149763421002700]
- 69. Precision diagnostic and therapeutic interventions in rare ... [https://www.nature.com/articles/s41390-025-04611-y]
- 70. The genetic landscape of autism spectrum disorder in an ... [https://www.nature.com/articles/s41525-024-00444-6]
- 71. A Guide to Cross-Validation for Artificial Intelligence in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10388213/]
- 72. Solving 9 Common Cross-Validation Mistakes [https://medium.com/@jan_marcel_kezmann/solving-9-common-cross-validation-mistakes-ac8a6a6944e7]
- 73. Cross validation – a safeguard for machine learning models [https://ardigen.com/cross-validation-a-safeguard-for-machine-learning-models/]
- 74. 3.1. Cross-validation: evaluating estimator performance [https://scikit-learn.org/stable/modules/cross_validation.html]
- 75. Whole genome sequencing analysis identifies sex ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11429951/]
- 76. Bridging genomics' greatest challenge: The diversity gap [https://www.sciencedirect.com/science/article/pii/S2666979X24003537]
- 77. Genomic architecture of autism from comprehensive whole ... [https://www.sciencedirect.com/science/article/pii/S0092867422013241]
- 78. New Autism Subtypes Identified Based on Genes, ... [https://www.brainfacts.org/diseases-and-disorders/childhood-disorders/2025/new-autism-subtypes-identified-based-on-genes-individual-traits-111125]
- 79. SLC35G1 is a highly chloride-sensitive transporter ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11542916/]
- 80. SLC35G1: A highly chloride-sensitive transporter ... [https://elifesciences.org/reviewed-preprints/98853]