A Researcher's Guide to PPI Data: Mastering IntAct, BioGRID, and Beyond for Robust Network Analysis
This guide provides researchers, scientists, and drug development professionals with a comprehensive framework for leveraging public Protein-Protein Interaction (PPI) databases.
A Researcher's Guide to PPI Data: Mastering IntAct, BioGRID, and Beyond for Robust Network Analysis
Abstract
This guide provides researchers, scientists, and drug development professionals with a comprehensive framework for leveraging public Protein-Protein Interaction (PPI) databases. It covers foundational knowledge of major resources like IntAct and BioGRID, strategic methodologies for data integration and tissue-specific application, solutions for common challenges including data heterogeneity and validation, and finally, best practices for comparative analysis and quality assessment. The article synthesizes current practices and emerging trends to empower the construction of biologically relevant PPI networks for advanced biomedical discovery.
Navigating the PPI Landscape: A Deep Dive into Core Databases and Their Specialties
Protein-protein interactions (PPIs) are fundamental to virtually every cellular process, from signal transduction and metabolic regulation to DNA replication and immune response. The systematic mapping of these interactions has become a cornerstone of modern biology, enabling researchers to model complex cellular networks and identify novel therapeutic targets. Public PPI databases have emerged as critical infrastructure for the life sciences, providing centralized, curated repositories of interaction data. These resources transform scattered experimental findings from the scientific literature into structured, computationally accessible knowledge. The field is characterized by a collaborative yet complementary ecosystem of databases, each with distinct strengths in curation focus, data types, and analytical tools. This guide provides an in-depth technical examination of six core resources—IntAct, BioGRID, HPRD, MINT, DIP, and REACTOME—framed within the context of biomedical research and drug discovery.
Database Comparison and Technical Specifications
The following table summarizes the key technical specifications and content focus of each major PPI database, enabling researchers to quickly identify the most appropriate resource for their specific needs.
Table 1: Core Features of Major Public PPI Databases
| Database | Primary Focus | Data Coverage | Curation Approach | Key Features |
|---|---|---|---|---|
| BioGRID | Protein & genetic interactions [1] [2] | ~1.93M interactions (2020); Human (670K), Yeast (755K) [2] | Manual curation from high & low-throughput studies [1] [2] | Includes PTMs, chemical interactions, CRISPR screens (ORCS) [2] |
| HPRD | Human proteome annotation [3] [4] | 20,000+ proteins; 30,000+ PPIs (2009) [3] | Manual literature extraction by biologists [3] [4] | PhosphoMotif Finder, disease associations, linked to NetPath [3] [4] |
| MINT | Experimentally verified PPIs [5] | Focused on curated physical interactions | Expert manual curation, PSI-MI standards [5] | IMEx consortium member; data integrated via IntAct [5] |
| DIP | Experimentally determined PPIs [6] | 1,089 proteins; 1,269 interactions (1999) [6] | Manual entry with expert review [6] | Details domains, amino acid ranges, dissociation constants [6] |
| IntAct | Molecular interaction data [7] | Provides molecular interaction data | Open source database; PSICQUIC service [7] | Confidence scores (MI score ≥ 0.45); framework for other resources [7] |
| REACTOME | Pathways & reactions [8] [9] | 2,825 human pathways; 16,002 reactions [8] | Manually curated, peer-reviewed pathways [9] | SBGN visualization; orthology-based predictions for 20 species [9] |
Table 2: Data Accessibility and Integration Features
| Database | Download Formats | Programmatic Access | Integration/Partnerships |
|---|---|---|---|
| BioGRID | Multiple formats including PSI MI XML [1] | REST API, Cytoscape plugin [1] | IMEx; data feeds to SGD, TAIR, FlyBase [1] |
| HPRD | Not specified | Human Proteinpedia submission portal [3] [4] | Linked to NetPath signaling pathways [3] [4] |
| MINT | PSI-MI standards [5] | PSICQUIC webservice [5] | IMEx consortium; data in IntAct [5] |
| DIP | Relational SQL database [6] | Web editing interface [6] | Links to sequence databases and pathway resources [6] |
| IntAct | Standardized downloads | PSICQUIC service [7] | Hosts MINT data; PSICQUIC aggregator [5] [7] |
| REACTOME | Various formats including SBGN [9] | Analysis tools API [8] | Overlays data from IntAct, BioGRID, MINT, etc. [9] |
Experimental Methodologies and Curation Standards
Curation Pipelines and Evidence Codes
PPI databases employ rigorous curation methodologies to ensure data quality and reliability. BioGRID maintains particularly detailed curation standards, with all interactions exclusively derived from manual curation of experimental data in peer-reviewed publications [2]. Each interaction is assigned structured evidence codes, including 17 different protein interaction evidence types (e.g., affinity capture-mass spectrometry, co-crystal structure, FRET, two-hybrid) and 11 genetic interaction evidence codes (e.g., synthetic lethality, synthetic rescue, dosage growth defect) [2]. This granular approach allows researchers to assess experimental context and reliability. High-throughput datasets are typically extracted from supplementary files and converted into consistent formats, while computationally predicted interactions are explicitly excluded to maintain high-confidence data standards [2].
Orthology-Based Pathway Predictions
REACTOME employs a sophisticated orthology inference system to extend human pathway knowledge to model organisms. The platform uses Ensembl Compara to identify orthologs of curated human proteins across 20 different species, enabling electronic inference of conserved reactions and pathways [9]. This approach significantly expands the utility of REACTOME for comparative biology and studies using model organisms. The Species Comparison tool allows direct comparison of predicted pathways between human and selected species, facilitating evolutionary analyses and translational research [9].
Data Integration Frameworks
The International Molecular Exchange (IMEx) consortium represents a critical collaborative framework in the PPI database ecosystem, with MINT and BioGRID as participating members [5] [2]. IMEx establishes common curation standards and enables resource sharing to minimize redundancy. The PSICQUIC (Proteomics Standard Initiative Common QUery InterfaCe) web service provides unified programmatic access to multiple interaction databases, including IntAct, BioGRID, MINT, and others [5] [9]. This interoperability allows researchers to query multiple resources simultaneously and facilitates more comprehensive network analyses.
Pathway Visualization and Data Integration
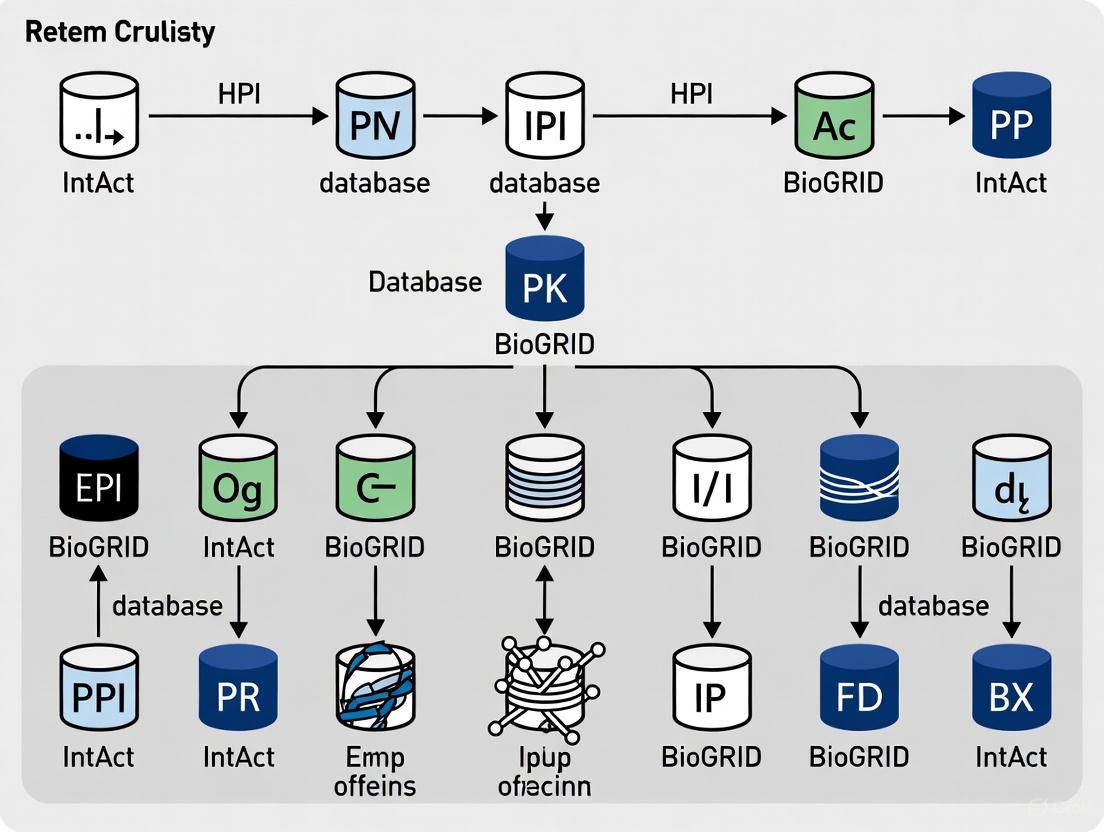
The following diagram illustrates the relationships and data integration between the major PPI databases and analytical tools:
Diagram 1: PPI Database Ecosystem and Data Flow (64 characters)
REACTOME's Pathway Browser implements Systems Biology Graphical Notation (SBGN) for standardized pathway visualization [9]. This enables consistent representation of biological entities and processes across different pathway diagrams. The browser supports zooming, scrolling, and event highlighting, with context-sensitive menus providing additional information about pathway components. A key innovation is the ability to overlay curated pathways with molecular interaction data from external databases, including IntAct, BioGRID, MINT, and others via PSICQUIC web services [9]. This integration creates a powerful environment for contextualizing interaction networks within established pathway frameworks.
Table 3: Key Research Reagent Solutions for PPI Studies
| Reagent/Resource | Function in PPI Research | Example Applications |
|---|---|---|
| CRISPR/Cas9 Systems | Gene knockout for genetic interaction screens | BioGRID-ORCS: 1,042+ CRISPR screens in human, mouse, fly [2] |
| Affinity Capture Reagents | Antibodies for immunoprecipitation | BioGRID evidence code: affinity capture-MS [2] |
| Two-Hybrid Systems | Binary interaction detection | Yeast two-hybrid; documented in DIP, BioGRID [6] [2] |
| Mass Spectrometry | Identification of co-purified proteins | Large-scale interaction datasets; PTM detection [2] [4] |
| PSICQUIC Tools | Unified querying of multiple databases | Programmatic access to IntAct, BioGRID, MINT [5] [9] |
The experimental workflow for generating and analyzing PPI data involves multiple complementary techniques, as shown in the following diagram:
Diagram 2: PPI Experimental and Analysis Workflow (52 characters)
Research Applications and Future Directions
Themed Curation for Disease-Focused Research
BioGRID has implemented themed curation projects to build depth in critical areas of human biology and disease [2]. These focused efforts include the ubiquitin-proteasome system (UPS), chromatin modification, autophagy, glioblastoma, Fanconi anemia, and most recently, SARS-CoV-2 coronavirus interactions [2]. Domain experts develop curated gene/protein lists to guide literature curation strategies, enabling comprehensive coverage of these specialized areas. This approach demonstrates how PPI databases can evolve beyond general repositories to become targeted discovery tools for specific research communities and disease areas.
Expanding Data Types and Modalities
The PPI database landscape continues to evolve beyond simple binary interactions. BioGRID now captures over 515,000 unique protein post-translational modifications and more than 28,000 interactions between drugs/chemicals and their protein targets [2]. The development of BioGRID-ORCS (Open Repository of CRISPR Screens) extends this further by capturing single mutant phenotypes and genetic interactions from genome-wide CRISPR/Cas9 screens [2]. This expansion reflects the growing integration of multi-modal data in network biology, providing richer context for interpreting interaction networks.
Challenges and Considerations
Researchers must recognize several considerations when using these resources. Data currency varies significantly between databases; for example, HPRD has not been updated since 2009, while BioGRID and REACTOME maintain regular updates [3] [8] [2]. Species coverage differs substantially, with some resources focusing exclusively on human data while others encompass multiple model organisms. Evidence quality should be critically evaluated through experimental method annotations and confidence scores. The complementary nature of these resources often necessitates querying multiple databases to obtain comprehensive interaction networks for a protein of interest.
Protein-protein interaction (PPI) data is fundamental to systems biology, providing critical insights into cellular signaling, regulatory pathways, and the molecular mechanisms underlying disease. For researchers, scientists, and drug development professionals, selecting the appropriate database is crucial for experimental design and data interpretation. This technical guide provides a comprehensive comparison of major PPI resources, focusing on their distinct curation methodologies, coverage, and specialized strengths to inform their use within biomedical research pipelines.
Key PPI Databases at a Glance
Table 1: Core Features of Major Protein-Protein Interaction Databases
| Database | Primary Focus | Curation Policy | Interaction Types | Notable Strengths |
|---|---|---|---|---|
| BioGRID [10] [11] | Protein, genetic, and chemical interactions for major model organisms and humans | Manual curation from literature; no unpublished data or reviews [12] | Physical, genetic, chemical, post-translational modifications | Extensive genetic interaction data; CRISPR screen data via ORCS [11] [13] |
| IntAct [14] | Molecular interaction data from literature curation and direct submissions | Open-source, open data; IMEx-level annotation and MIMIx-compatible entries [14] | Protein-protein, protein-small molecule, protein-nucleic acid | Detailed experimental condition description; compliant with IMEx consortium standards [14] |
| APID [15] | Unified "interactomes" by integrating data from primary sources | Data integration and unification from primary databases (e.g., BioGRID, IntAct, HPRD, MINT, DIP) [15] | Protein-protein (with "binary" vs "indirect" classification) | Provides unified, non-redundant interactomes; distinguishes binary physical interactions [15] |
| STRING [16] | Experimental and predicted interactions | Integration of curated data and predictions from genomic context, text-mining, etc. [17] | Experimental and predicted | High coverage; combined results with UniHI cover ~84% of experimentally verified PPIs [16] |
Quantitative Coverage and Performance
A systematic comparison of 16 PPI databases provides critical metrics for database selection based on coverage. The study found that combined results from STRING and UniHI covered approximately 84% of 'experimentally verified' PPIs for a test set of genes. For 'total' interactions (including predicted), about 94% of available PPIs were retrieved by the combined use of hPRINT, STRING, and IID. Among exclusively found experimentally verified PPIs, STRING contributed around 71% of the unique hits. Analysis with a gold-standard set of curated interactions revealed that GPS-Prot, STRING, APID, and HIPPIE each covered approximately 70% of these high-quality interactions [16].
Table 2: Database Coverage Metrics from a User's Perspective Study [16]
| Metric | Finding | Key Databases |
|---|---|---|
| Experimentally Verified PPIs | ~84% coverage | Combined use of STRING & UniHI |
| Total PPIs (Experimental & Predicted) | ~94% coverage | Combined use of hPRINT, STRING, & IID |
| Exclusively Found PPIs | ~71% of unique hits | STRING |
| Gold-Standard Curated PPIs | ~70% coverage each | GPS-Prot, STRING, APID, HIPPIE |
Specialized databases have also been developed for specific biological contexts. For instance, InterMitoBase, a database for human mitochondrial PPIs, contains 5,883 non-redundant interactions from 2,813 proteins integrated from PubMed, KEGG, BioGRID, HPRD, DIP, and IntAct. Of these, 1,640 are novel interactions not covered by the four major PPI databases [18].
Detailed Curation Methods and Workflows
BioGRID Curation Methodology
BioGRID employs a rigorous manual curation process where all interactions are captured as gene identifier pairs from the primary literature. The curation workflow involves:
- Evidence Capture: Curators record interacting partners, experimental evidence codes, and PubMed IDs. They capture all interactions in a paper, including those not the main focus and interactions found in supplementary files [10].
- Experimental Classification: Interactions are categorized using controlled vocabularies mapping to the PSI-MI 2.5 standard. Key distinctions include:
- Quality Assurance: Interactions from retracted publications are systematically removed, though data conflicts between non-retracted publications are maintained as a reflection of the literature [10].
- CRISPR Screen Curation: Through its ORCS (Open Repository for CRISPR Screens) platform, BioGRID curates genome-wide screens, capturing gene-phenotype and gene-gene relationships, including cell line metadata, experimental conditions, and gRNA library information [11] [13].
The following workflow diagram illustrates BioGRID's comprehensive curation process:
IntAct Curation and Quality Standards
IntAct employs a dual-level curation system with stringent quality control measures:
- Curation Standards: Supports both IMEx-level annotation (comprehensive details of experimental conditions, constructs, and participant methodologies) and MIMIx-compatible entries (less comprehensive but capturing essential data for confidence assessment) [14].
- Participant Detail: Protein interactions can be described to the isoform level or post-translationally cleaved mature peptide level using appropriate UniProtKB identifiers. Participant status is checked with every UniProtKB release, with remapping performed when sequences are updated or withdrawn [14].
- Quality Control: Each entry undergoes peer review by a senior curator before release. Additional rule-based checks are run at the database level, and original authors are contacted to verify data representation after release [14].
- Interaction Scoring: IntAct employs a quantitative scoring system for exporting interactions to UniProtKB/GOA, weighting interaction detection methods (e.g., Biochemical=3, Biophysical=3) and interaction types (e.g., Direct interaction=5, Association=1) [14].
APID Data Integration and Redefinition
APID functions as a meta-database that redefines and unifies PPI data from primary sources through a systematic pipeline:
- Data Unification: Integrates PPIs from BioGRID, DIP, HPRD, IntAct, and MINT, plus human data sources and 3D structures from PDB, removing duplicate and incomplete records [15].
- Method Re-evaluation: APID re-evaluated PSI-MI ontological vocabulary to distinguish proper experimental methods, creating a new "Method_Type" category with 11 terms labeled "NotAssigned" as they don't demonstrate experimental detection [15].
- Binary vs. Indirect Classification: APID classifies interaction detection methods as "binary" (direct physical detection between specific protein pairs) or "indirect" (detecting interactions within protein groups without direct pairwise distinction) [15].
- Binary Interactomes: This classification enables construction of binary interactomes, with the human binary interactome containing 83,949 PPIs (~21% of all reported human interactions) but covering over 90% of the reference proteome [15].
The following diagram illustrates APID's data integration and refinement pipeline:
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents and Resources for PPI Research
| Resource/Reagent | Function in PPI Research | Application Context |
|---|---|---|
| CRISPR/Cas9 gRNA Libraries | Genome-wide screening for gene-phenotype and gene-gene relationships [11] | Identification of novel genetic interactions and functional gene modules |
| Affinity Tags (TAP, GST, etc.) | Protein purification and interaction capture for mass spectrometry or western analysis [10] | In vivo and in vitro interaction validation (Affinity Capture-MS/Western) |
| PSI-MI Controlled Vocabularies | Standardized annotation of experiments for consistent data interchange [15] | Database curation, data sharing, and meta-analysis across resources |
| Antibodies for Immunoblotting | Detection of specific proteins in co-immunoprecipitation experiments [10] | Validation of physical interactions and complex formation |
| Recombinant Protein Expression Systems | Production of purified proteins for in vitro interaction studies [10] | Reconstituted complex experiments and direct binding assays |
The landscape of PPI databases offers diverse resources with complementary strengths. BioGRID excels in genetic interactions and manual curation from literature, IntAct provides exceptionally detailed experimental annotations adhering to IMEx consortium standards, APID offers unified, non-redundant interactomes distinguishing binary interactions, and STRING delivers broad coverage by integrating experimental and predicted data. Research indicating that database usage frequencies do not always correlate with their respective advantages underscores the importance of informed selection [16]. For researchers in drug development and biomedical science, strategic use of multiple databases—particularly those with complementary coverage—provides the most comprehensive foundation for network analysis and therapeutic discovery.
The Biological General Repository for Interaction Datasets (BioGRID) is a primary database for the collection and standardization of protein-protein and genetic interactions. Its mission is to provide a comprehensive repository of molecular interactions that are manually curated from the primary biomedical literature, enabling systems-level biological approaches and facilitating the understanding of human disease and physiology. Unlike computationally predicted interactions, BioGRID provides experimentally evidenced data, making it an essential resource for researchers validating disease targets, understanding signaling pathways, and building network models of cellular processes. The core principle of BioGRID's curation philosophy is the systematic capture of binary molecular relationships directly supported by experimental evidence, providing researchers with a reliable foundation for network analysis and hypothesis generation [12] [19]. This technical guide details the principles, workflow, and methodologies underlying BioGRID's publication-driven curation process, providing researchers with the contextual knowledge needed to effectively utilize this critical bioinformatics resource.
Core Curation Principles and Data Scope
BioGRID operates on several foundational principles that govern what data is curated and how it is represented. Understanding these principles is essential for properly interpreting the interaction data provided by the resource.
- Publication-Driven Evidence: BioGRID curators exclusively capture interactions from primary research articles that provide direct experimental evidence. The database does not include interactions reported only in reviews or as unpublished data, ensuring that all curated interactions are traceable to a verifiable, peer-reviewed source [12].
- Binary Relationship Model: All interactions are recorded as binary relationships between two genes or proteins, accompanied by an evidence code that describes the experimental system used to detect the interaction and a reference to the supporting publication. This standardized format ensures computational tractability and consistent data representation [20].
- Comprehensive Organism Coverage: While BioGRID aims to curate all interactions for major model organisms, it also selectively curates topic-driven human datasets relevant to specific diseases or biological processes. The database encompasses interactions from numerous organisms, with substantial data for Homo sapiens, Saccharomyces cerevisiae, Drosophila melanogaster, and Arabidopsis thaliana, among others [21].
- Evidence over Interpretation: BioGRID curates interaction evidence as presented in each publication independently, without making judgments on the absolute biological truth of an interaction. This approach can lead to data conflicts in the database, which simply reflect contradictions in the published literature itself [10].
Table 1: BioGRID Data Statistics (Latest Build 4.4.241 - January 2025)
| Organism | Physical Interactions (Non-Redundant) | Genetic Interactions (Non-Redundant) | Unique Publications |
|---|---|---|---|
| Homo sapiens | 1,009,107 | 18,689 | 39,579 |
| Saccharomyces cerevisiae | 268,815 | 424,370 | 9,811 |
| Drosophila melanogaster | 68,703 | 10,764 | 8,053 |
| Arabidopsis thaliana | 74,009 | 299 | 2,450 |
| Caenorhabditis elegans | 41,075 | 2,295 | 1,560 |
The Curation Workflow: From Publication to Database
The BioGRID curation process follows a systematic workflow designed to ensure consistency and accuracy across all curated data. The workflow can be visualized as a multi-stage process where curators extract specific information from scientific publications and record it in a standardized format.
Diagram 1: BioGRID Curation Workflow
Literature Screening and Selection
The curation process begins with the identification of relevant scientific publications that contain reportable interaction data. BioGRID employs multiple strategies for literature identification, including automated PubMed searches, direct author submissions, and monitoring of high-impact journals. Curators prioritize articles that report novel interactions while also capturing additional evidence for previously reported interactions from new publications. The database focuses on comprehensive curation of all interactions within a paper, even those not central to the main findings or previously curated, to build a complete evidence trail for each interaction [10].
Interaction Identification and Data Extraction
Once a publication is selected for curation, expert curators perform a detailed reading of the full text to identify all reportable interactions. During this phase, curators:
- Identify specific figures, tables, and results sections that provide experimental evidence for molecular interactions
- Distinguish between physical interactions (direct binding or co-complex membership) and genetic interactions (functional relationships between genes)
- Extract the specific genes/proteins involved in each interaction, using standardized gene identifiers
- Determine experimental context, including the organism, cell type, and specific experimental conditions [19]
For complex data sets, particularly those from high-throughput studies presented in supplementary tables, curators may employ specialized loading scripts to efficiently process large numbers of interactions while maintaining data quality [10].
Experimental Evidence Annotation
A critical component of BioGRID curation is the annotation of the experimental evidence supporting each interaction. The database employs a detailed evidence code system that precisely describes the experimental methodology used to detect each interaction. This system allows users to assess the nature and quality of evidence supporting any given interaction in the database [20].
For each experiment supporting an interaction, curators record:
- The specific experimental system from the standardized evidence code ontology
- The publication reference (PubMed ID)
- The interacting partners as a pair of gene identifiers
- For physical interactions, directionality (bait and prey) when clearly defined in the experiment
- Any relevant qualifications or experimental details in free-text form [12]
Data Standardization and Quality Control
Before integration into the public database, all curated interactions undergo standardization and quality control checks. This process includes:
- Identifier standardization: Ensuring all interactors use correct and consistent gene identifiers across the database
- Evidence code validation: Verifying that the assigned evidence codes accurately reflect the described experimental methodology
- Directionality checks: Applying consistent rules for bait-prey relationships in pull-down experiments and other directional assays
- Duplicate detection: Identifying and appropriately handling interactions that may have been reported in multiple publications
BioGRID employs a spoke model for representing interactions, where a bait protein is connected to all identified prey proteins. This avoids artificial inflation of interaction counts that can occur when reciprocally validating interactions [10].
Database Integration and Release
The final stage of the curation workflow involves integrating the curated data into the BioGRID database and making it publicly available through regular quarterly releases. The database provides multiple access methods, including:
- A web-based search interface for interactive querying
- Bulk download files in standard formats for computational analysis
- A web service (API) for programmatic access to the data
- Specialized themed projects focused on specific biological processes or diseases [22]
Experimental Evidence Codes: A Detailed Taxonomy
BioGRID employs a comprehensive classification system for experimental evidence that enables precise annotation of the methods used to detect each interaction. This detailed taxonomy allows users to filter interactions based on experimental approach and assess the nature of supporting evidence.
Physical Interaction Evidence Codes
Physical interaction evidence codes describe experimental systems that detect direct or indirect physical associations between molecules. The specific methodologies are categorized as follows:
Table 2: Physical Interaction Evidence Codes in BioGRID
| Evidence Code | Experimental Principle | Key Methodological Features |
|---|---|---|
| Affinity Capture-MS | Protein complex isolation followed by mass spectrometry | Bait protein affinity-captured from cell extracts; associated partners identified by MS [20] |
| Affinity Capture-Western | Protein complex isolation followed by immunoblotting | Bait affinity-captured; interaction partners identified by Western blot with specific antibodies [20] |
| Co-crystal Structure | Direct atomic-level demonstration of interaction | X-ray crystallography, NMR, or EM structures showing physical interaction at atomic resolution [20] |
| Two-hybrid | Protein interaction detection via reporter gene activation | Bait expressed as DBD fusion, prey as TAD fusion; interaction measured by reporter activation [20] |
| FRET | Detection of molecular proximity by energy transfer | Fluorescence resonance energy transfer between fluorophore-labeled molecules in live cells [20] |
| Reconstituted Complex | In vitro demonstration of interaction between purified components | Includes GST pull-downs, surface plasmon resonance, bio-layer interferometry with recombinant proteins [20] [10] |
| Proximity Label-MS | Enzymatic labeling of vicinal proteins followed by MS | BioID and similar systems; bait-enzyme fusion labels nearby proteins for capture and identification [20] |
Genetic Interaction Evidence Codes
Genetic interactions describe functional relationships between genes, typically revealed through combinatorial genetic perturbations. Key genetic evidence codes include:
- Synthetic Lethality: Double mutant combination results in lethality, while single mutants are viable [10]
- Dosage Lethality: Overexpression of one gene causes lethality in a mutant background of another gene [20]
- Dosage Growth Defect: Overexpression causes a growth defect in a mutant background [20]
- Phenotypic Suppression: Mutation or overexpression of one gene reverses the phenotypic effect of another mutation
- Synthetic Rescue: Mutation in one gene reverses the phenotypic effect of another mutation [10]
BioGRID curators capture genetic interactions only when single mutants and double/multiple mutants are directly compared within the same publication or clearly referenced, ensuring the reliability of the genetic interaction evidence [10].
Distinguishing Between Similar Experimental Systems
Curatorial judgment is particularly important for distinguishing between experimentally similar but conceptually distinct evidence codes. Key differentiations include:
- Affinity Capture vs. Reconstituted Complex: The critical distinction is whether the relevant proteins are co-expressed in the cell (Affinity Capture) or the interaction is demonstrated in vitro with purified components (Reconstituted Complex) [10]
- Affinity Capture-Western/MS vs. Co-purification: Co-purification involves at least one extra purification step beyond standard affinity capture methods to remove potential contaminating proteins [20]
- Protein-RNA vs. Affinity Capture-RNA: Protein-RNA covers in vitro interactions, while Affinity Capture-RNA involves protein and RNA co-expressed in vivo [20]
Specialized Curation Projects and Methodologies
Beyond its core curation activities, BioGRID has developed specialized curation projects and methodologies to address specific biological questions and data types.
Themed Curation Projects
BioGRID's themed curation projects focus on specific biological processes with disease relevance. These projects involve:
- Assembling core genes/proteins central to a biological process with expert input
- Curating relevant publications for biological interactions with enhanced annotation
- Providing focused datasets for diseases including Alzheimer's Disease, COVID-19 Coronavirus, Autism Spectrum Disorder, and Glioblastoma [22]
These themed projects are updated monthly and provide researchers with pre-compiled interaction networks for specific pathological contexts.
BioGRID ORCS: CRISPR Screen Curation
The BioGRID Open Repository of CRISPR Screens (ORCS) is a specialized database for CRISPR screen data compiled through comprehensive curation of genome-wide CRISPR screens reported in the literature. ORCS provides:
- Structured metadata annotation capturing salient CRISPR experimental details
- Data from hundreds of publications encompassing thousands of individual screens
- Search functionality by gene/protein, phenotype, cell line, and other attributes [22]
Handling Complex Curation Scenarios
BioGRID curators follow specific guidelines for handling complex or edge-case scenarios:
- Self-interactions: Recorded when clear evidence exists for homodimerization or multimerization, most commonly through tagging a single protein with two different tags and demonstrating interaction [10]
- Cross-species interactions: Curated between proteins from different species, excluding cross-species complementation experiments that test functional orthology rather than genetic interaction [10]
- "Data not shown" interactions: Generally not curated, but curator judgment may be applied when the interactions are clearly supported by the experimental context [10]
- Conflicting data: All interactions are curated as presented in each publication, reflecting the current state of the literature, with systematic removal only for retracted publications [10]
Research Reagent Solutions for Interaction Studies
The experimental methods captured by BioGRID evidence codes rely on specific research reagents and tools. The table below details key reagents and their applications in interaction studies.
Table 3: Essential Research Reagents for Interaction Studies
| Research Reagent | Primary Function | Application in Interaction Studies |
|---|---|---|
| Epitope Tags (TAP, HA, FLAG) | Protein labeling and detection | Enable affinity capture of bait proteins and their interaction partners [20] |
| Polyclonal/Monoclonal Antibodies | Target-specific protein recognition | Used for Western blot detection and immunoprecipitation in affinity capture experiments [20] |
| Luciferase Reporters | Bioluminescence detection | Serve as detectable markers in protein-fragment complementation assays [20] |
| Fluorescent Proteins (CFP, YFP) | Fluorescence emission | Act as donor-acceptor pairs in FRET-based interaction detection [20] |
| Cross-linking Reagents | Covalent protein linkage | Stabilize transient interactions for Cross-Linking-MS studies [20] |
| GST Fusion Systems | Affinity purification | Facilitate pull-down assays for Reconstituted Complex experiments [20] [10] |
| CRISPR Libraries | Gene knockout screening | Enable genome-wide functional genetic interaction studies [22] |
Data Access and Utilization
Access Methods and Formats
BioGRID provides multiple access pathways to accommodate diverse research needs:
- Web Interface: Interactive searching with filters for organisms, interaction types, and evidence codes
- Bulk Downloads: Complete datasets available in standard formats including MITAB, PSI-MI, and BioPLEX
- Web Services: RESTful API access for programmatic querying and integration into analytical pipelines
- Browser Extensions: GIX extension retrieves gene product information directly on any webpage by double-clicking gene names [22]
Data Statistics and Growth
As of the latest 2025 statistics, BioGRID has curated interactions, chemical associations, and post-translational modifications from over 87,000 publications. The database contains:
- More than 2.25 million non-redundant molecular interactions
- Over 14,000 non-redundant chemical associations
- Nearly 564,000 non-redundant post-translational modification sites [22]
The database undergoes monthly curation updates, with new data added on a continuous basis to maintain current coverage of the scientific literature.
BioGRID data interoperates with numerous complementary resources through data sharing and standardization initiatives:
- STRING Database: BioGRID physical and genetic interactions contribute to the evidence network in STRING, which integrates additional prediction algorithms and pathway databases [23]
- Pathway Databases: Interactions from BioGRID feed into Reactome, KEGG, and other pathway resources
- Model Organism Databases: Collaboration with organism-specific databases ensures consistent gene annotation and identifier mapping
BioGRID's publication-driven curation model provides an essential foundation for systems biology and network-based approaches to understanding cellular function and disease mechanisms. By manually extracting experimentally supported interactions from the literature and representing them in a standardized, computationally accessible format, BioGRID enables researchers to move beyond individual interactions to system-level analyses. The detailed annotation of experimental evidence allows users to assess the nature and quality of support for each interaction, while the comprehensive coverage across model organisms and human datasets facilitates comparative network biology. As the volume of interaction data continues to grow, BioGRID's rigorous curation standards and specialized projects will remain critical for distilling high-quality molecular interaction networks from the expanding biomedical literature.
The Critical Role of Manual Curation and Expert Review in Databases like DIP and HPRD
In the complex landscape of systems biology, protein-protein interaction (PPI) networks serve as fundamental maps for understanding cellular processes and disease mechanisms. The accuracy and reliability of these networks depend critically on the curation processes behind the databases that house them. Manual curation and expert review represent the gold standard in this field, transforming raw experimental data into biologically meaningful information. Databases such as the Human Protein Reference Database (HPRD) and the Database of Interacting Proteins (DIP) have established themselves as authoritative resources precisely because of their rigorous curation methodologies. These curated databases form the foundation for diverse biomedical applications, from identifying novel drug targets to understanding the molecular basis of genetic diseases. Within the broader ecosystem of PPI resources that includes repositories like IntAct and BioGRID, the distinctive value of manually curated databases lies in their ability to provide context, resolve contradictions, and maintain consistently high-quality annotations across the entire proteome.
The essential challenge in PPI database management stems from the tremendous heterogeneity in experimental data quality and methodology. As Cusick et al. noted, different experimental techniques—from yeast two-hybrid (Y2H) systems to affinity purification followed by mass spectrometry (AP-MS)—produce fundamentally different types of interaction data [24]. Without expert interpretation, these data remain isolated facts rather than connected biological knowledge. Manual curation addresses this limitation by applying consistent standards and biological expertise to create structured, searchable, and interconnected data resources. This whitepaper examines the critical curation methodologies, quantitative impacts, and practical applications of manual curation in PPI databases, providing researchers with a comprehensive framework for leveraging these essential resources.
Manual Curation Methodologies: Protocols and Workflows
Core Curation Workflow
The manual curation process in databases like HPRD and DIP follows a systematic protocol to ensure consistency and accuracy. The workflow begins with comprehensive literature surveillance, where curators identify relevant publications containing experimental protein interaction data. This initial screening process typically employs sophisticated text-mining algorithms to identify candidate papers, which are then subjected to expert biological review. Trained curators, often holding advanced degrees in molecular biology or related fields, carefully examine the experimental details, methodology, and results reported in each publication.
The critical evaluation phase involves assessing the experimental evidence according to predefined quality metrics. Curators extract essential information including the specific experimental method used (e.g., Y2H, co-immunoprecipitation, TAP-MS), experimental conditions, interaction domains identified, and any quantitative measurements of binding affinity. This information is then structured according to standardized ontologies, particularly the Proteomics Standards Initiative - Molecular Interaction (PSI-MI) format, which enables data exchange and integration across resources [24]. Throughout this process, curators make critical judgments about which interactions meet quality thresholds for inclusion, resolving ambiguities in the primary literature that automated methods might overlook.
Figure 1: The sequential workflow for manual curation of protein-protein interaction data, highlighting the stages from literature identification to public release.
Specialized Curation Protocols for Different Experimental Methods
Manual curation requires distinct approaches for different experimental methodologies. For yeast two-hybrid experiments, curators focus on validating the binary nature of interactions, examining bait-prey pairs, and assessing false-positive rates based on control experiments. For affinity purification-mass spectrometry approaches, curators face the additional complexity of distinguishing direct physical interactions from co-purifying components of protein complexes. In this context, the curation protocol must address the representation model—whether to use the "matrix" model (assuming all components interact with each other) or the "spokes" model (connecting the bait protein to each prey) [24].
HPRD has developed particularly sophisticated curation protocols for post-translational modifications (PTMs), with phosphorylation events constituting 63% of all PTM data in the database [25]. For these annotations, curators not only record the modification itself but also contextual information including the modifying enzyme, specific modified residues, and functional consequences of the modification. This granular level of detail enables researchers to construct regulatory networks that extend beyond simple physical interactions to include functional relationships. The PhosphoMotif Finder tool within HPRD further exemplifies specialized curation, containing known kinase/phosphatase substrate and binding motifs curated exclusively from published literature [25].
Quantitative Impact of Manual Curation on Data Quality and Coverage
Coverage Statistics of Major PPI Databases
The rigorous manual curation methodologies employed by databases like HPRD and DIP directly translate into superior data quality and unique coverage advantages. The table below summarizes the documented coverage of major PPI databases, highlighting the distinctive position of manually curated resources:
Table 1: Protein-Protein Interaction Database Coverage Comparisons
| Database | Primary Curation Method | Reported Interactions | Publication Sources | Organism Focus | Key Strengths |
|---|---|---|---|---|---|
| HPRD | Manual expert curation | 38,000+ PPIs (2009) [26] | 18,777+ publications [24] | Human-specific | Integrated PTM data, disease associations, tissue expression |
| DIP | Manual curation with binary interaction focus | 53,431 interactions (2008) [24] | 3,193 publications [24] | Multiple organisms (134 species) | High-quality binary interactions, IMEx consortium member |
| BioGRID | Mixed curation approaches | 42,800 human PPIs (2009) [26] | 16,369 publications (2008) [24] | Multiple organisms (10 species) | Extensive genetic interaction data, themed curation projects |
| IntAct | Mixed curation approaches | 129,559 interactions (2008) [24] | 3,166 publications [24] | Multiple organisms (131 species) | IMEx consortium partner, comprehensive species coverage |
| MINT | Mixed curation approaches | 80,039 interactions (2008) [24] | 3,047 publications [24] | Multiple organisms (144 species) | Confidence scoring, protein-promoter/mRNA interactions |
The quantitative evidence demonstrates that HPRD's manual curation approach enables coverage of substantially more scientific publications than other databases—over 18,000 publications compared to approximately 3,000 for several other resources [24]. This extensive literature mining translates into more comprehensive annotation of biologically relevant interactions, particularly those reported in smaller-scale studies that might be missed by approaches focusing primarily on high-throughput datasets.
Comparative Analysis of Interaction Overlap and Unique Contributions
Systematic comparisons reveal limited overlap between different PPI databases, with each resource contributing unique interactions. A study analyzing 14,899 publications shared across multiple databases found that 39% were reported with different numbers of interactions in different databases [24]. These discrepancies arise from varying curation standards, identifier mapping challenges, and different interpretations of experimental results. In one notable example, the same publication reporting human PPIs was documented with 2,371 interactions in HPRD, 2,671 in IntAct, and 2,463 in MINT, while BioGRID reported 6,295 interactions from the same study, indicating fundamental differences in curation methodology [24].
Manual curation particularly excels in capturing interactions from small-scale, hypothesis-driven studies that provide crucial biological context. Analysis has shown that combined use of STRING and UniHI covers approximately 84% of experimentally verified PPIs, while nearly 94% of total PPIs (experimental and predicted) require combined data from hPRINT, STRING, and IID [16]. However, these metrics of breadth must be balanced against quality assessments, with studies revealing that GPS-Prot, STRING, APID, and HIPPIE each cover approximately 70% of curated interactions from a gold-standard PPI set [16].
Key Research Reagent Solutions for PPI Investigation
Table 2: Essential Research Reagents and Resources for Protein-Protein Interaction Studies
| Resource/Reagent | Function/Application | Database Implementation |
|---|---|---|
| Yeast Two-Hybrid (Y2H) Systems | Detection of binary protein interactions | HPRD, DIP, BioGRID categorize Y2H-derived interactions with specific evidence tags |
| Tandem Affinity Purification (TAP) Tags | Protein complex purification for mass spectrometry | Curators distinguish bait-prey relationships in AP-MS data |
| Co-immunoprecipitation (Co-IP) Antibodies | Validation of physical interactions in native cellular environments | HPRD documents specific antibodies used in validated interactions |
| CRISPR Screening Libraries | Genome-wide functional interaction studies | BioGRID ORCS database compiles CRISPR screen data [22] |
| Phospho-Specific Antibodies | Detection of post-translational modifications | HPRD curates phosphorylation sites with modifying enzyme data |
| Proteomic Standards Initiative MI (PSI-MI) | Data standardization and exchange format | IMEx consortium databases (DIP, IntAct, MINT) use PSI-MI for data sharing [24] |
The specialized reagents and resources listed in Table 2 represent critical tools for generating experimentally validated PPI data. Manual curation databases document the specific experimental methods and reagents used to identify each interaction, enabling researchers to assess the reliability of specific data points. This granular documentation is particularly valuable when designing follow-up experiments, as it provides insight into validated experimental approaches.
Complementary Roles in the PPI Data Ecosystem
Manually curated databases like HPRD and DIP do not exist in isolation but function as crucial components within a broader ecosystem of PPI resources. Meta-databases such as STRING, UniHI, and APID aggregate data from multiple sources, including manually curated databases, to provide more comprehensive coverage [26] [27]. The distinct value of manually curated databases in this ecosystem lies in their role as authoritative sources for high-quality, context-rich interaction data. The integration relationships between these resources can be visualized as follows:
Figure 2: Integration framework showing how manually curated databases contribute to meta-databases and directly support research applications.
The critical importance of manual curation becomes evident when examining how these integrated resources are employed in practice. For example, STRING incorporates PPI information from HPRD, BioGRID, MINT, BIND, and DIP, and supplements these data with text-mining results and predicted interactions [26]. Similarly, UniHI integrates PPIs from both high-throughput yeast two-hybrid screens and curated databases including HPRD, DIP, BIND, and Reactome [26]. In these contexts, the manually curated data from HPRD and DIP serve as benchmark datasets for validating computational predictions and text-mining results.
Applications in Disease Research and Drug Development
From Network Biology to Therapeutic Insights
The rigorous manual curation practices employed by databases like HPRD directly enable important applications in disease research and drug development. The annotation of disease-associated proteins and their interconnection within PPI networks provides a systems-level framework for understanding pathogenesis. For example, HPRD explicitly links proteins involved in human diseases to the Online Mendelian Inheritance in Man (OMIM) database, creating a critical bridge between genetics and proteomics [25].
A compelling example of how manually curated PPI data advance disease research comes from a study of inherited neurodegenerative disorders characterized by ataxia. Lim et al. constructed a protein interaction network for 54 proteins involved in 23 ataxias by combining yeast two-hybrid data with literature-curated interactions from BIND, HPRD, DIP, and MINT [26]. This integrated network revealed unexpected connections between ataxia proteins, suggesting shared pathways and disease mechanisms that had not been apparent from studying individual proteins in isolation. The manually curated interactions were essential for establishing the biological relevance of the network, with 68% of literature-curated interactions and 63% of interlog interactions annotated to similar Gene Ontology compartments [26].
Manual curation also plays a crucial role in drug target identification and validation. By mapping disease-associated proteins within the broader context of interaction networks, researchers can identify critical hubs or bottlenecks that represent attractive therapeutic targets. The annotation of enzyme-substrate relationships in HPRD further supports drug discovery by identifying potential modulators of pathway activity [25]. For drug development professionals, these curated networks provide insight into potential mechanism-based toxicities and off-target effects by revealing unanticipated connections between pathways.
Future Directions and Implementation Recommendations
Advancing Curation Practices in the Era of Big Data Biology
As the volume and complexity of proteomic data continue to grow, manual curation methodologies must evolve to maintain their critical role in ensuring data quality. Future developments will likely involve more sophisticated human-computer partnership approaches, where expert curators train machine learning algorithms to handle routine annotation tasks while focusing their expertise on particularly complex or contradictory findings. The continued development and adoption of community standards through initiatives like IMEx and PSI-MI will be essential for enabling data integration while preserving the nuanced contextual information that manual curation provides [24].
For researchers and drug development professionals leveraging PPI data, we recommend a stratified approach to database selection and use. For initial exploratory network analysis, meta-databases like STRING and UniHI provide valuable comprehensive overviews. However, for hypothesis-driven research and validation studies, direct consultation of manually curated databases like HPRD and DIP is essential. When designing follow-up experiments, researchers should pay particular attention to the experimental methods documented in these curated resources, as they provide validated approaches for confirming specific interaction types. The continued support and utilization of manually curated databases will be essential for ensuring that our maps of the human interactome remain both comprehensive and biologically accurate.
Protein-protein interaction (PPI) data is fundamental to understanding cellular functions, with direct implications for drug discovery and the understanding of disease mechanisms. Resources like BioGRID and IntAct provide critical repositories of curated interaction data, making them indispensable for researchers in biomedical science [28]. However, the practical utility of these resources depends significantly on a researcher's ability to effectively access and utilize their data through various download formats and web interfaces. This guide provides a comprehensive technical overview of these access modalities, framed within the context of a broader thesis on PPI data resources. For researchers, scientists, and drug development professionals, selecting the appropriate data format and understanding access methodologies is not merely a preliminary step but a critical determinant of research efficiency and analytical success. The following sections detail the specific technical characteristics of major PPI databases, present structured comparisons, and provide actionable protocols for data retrieval and application.
The Biological General Repository for Interaction Datasets (BioGRID) is a comprehensive curated database of protein, genetic, and chemical interactions. As of late 2025, BioGRID release 5.0.251 contains curated data from over 87,393 publications, encompassing approximately 2.25 million non-redundant interactions and over 563,000 post-translational modification sites [29] [22]. This extensive repository is 100% freely available to both academic and commercial users under the MIT License, supporting open science initiatives without warranty restrictions [29] [30]. BioGRID's data is compiled through rigorous manual curation from the scientific literature, with updates released on a monthly basis to ensure researchers have access to the most current interaction information [22].
The IntAct Molecular Interaction Database is an open-source, open data resource maintained by the European Bioinformatics Institute (EBI). As a core member of the International Molecular Exchange (IMEx) consortium, IntAct provides fine-grained molecular interaction data curated from both scientific literature and direct data depositions [31]. The database employs a deep annotation model that captures extensive experimental details essential for the accurate interpretation of molecular interaction data. This granular approach to data curation ensures that researchers have access to the contextual experimental information necessary for robust biological conclusions. The IntAct platform also serves as a shared curation and dissemination platform for multiple global partners within the IMEx consortium, enhancing data standardization and accessibility [31].
Table 1: Core PPI Database Profiles
| Database | Primary Focus | Data Volume | Update Frequency | Licensing |
|---|---|---|---|---|
| BioGRID | Protein, genetic and chemical interactions | 2.25M+ non-redundant interactions from 87K+ publications [22] | Monthly [22] | MIT License [29] |
| IntAct | Molecular interactions with fine-grained annotation | 1M+ binary interactions (as of 2021) [31] | Regularly updated | Open source, open data [31] |
BioGRID Data Access Formats
Recommended File Formats
BioGRID provides data in multiple file formats, each designed for specific use cases and analytical workflows. For new projects, the following formats are recommended:
- PSI-MI XML 2.5 (PSI25): This standardized format follows the Proteomics Standards Initiative guidelines and is particularly suitable for data exchange and integration with other bioinformatics tools. The files contain extensive metadata about interactions and experimental conditions [32].
- BioGRID TAB 3.0 (TAB3): A tab-delimited format that offers a balance between comprehensive data content and ease of use. This format is particularly accessible for researchers using scripting languages like Python or R for data analysis, and it includes all core interaction information in a structured columnar format [32].
- Osprey Custom Network 1.3.1 (OSPREY): Specifically designed for network visualization and analysis in the Osprey Network Visualization System. This format optimizes data for graphical representation and topological analysis of interaction networks [32].
- PSI MITAB Version 2.5: A simplified tabular variant of the PSI-MI standard that facilitates easy parsing and processing while maintaining standardized interaction data representation [32].
Specialized Data Files
Beyond general interaction data, BioGRID offers several specialized datasets:
- Multi-Validated (MV) Physical Datasets: These files contain interactions that have been experimentally validated through multiple independent methods or publications, providing a high-confidence subset of physical interactions for rigorous analysis [29] [32].
- Chemical Interaction Data (CHEMTAB): This format captures bioactive chemical-protein relationships, including chemical perturbations and interactions, which is particularly valuable for drug discovery and chemical biology research [29] [32].
- Post-Translational Modification Data (PTM): These files provide comprehensive information on post-translational modification sites, a critical regulatory layer in cellular signaling pathways [29] [22].
- Themed Project Datasets: Focused datasets on specific disease areas or biological processes, including Alzheimer's disease, COVID-19 coronavirus, autism spectrum disorder, and glioblastoma [29] [22].
Table 2: BioGRID Download Formats and Specifications
| Format Type | File Extension | Typical Size Range | Primary Use Case |
|---|---|---|---|
| PSI-MI XML 2.5 | .psi25.zip | 181-200 MB | Data exchange, computational analysis [29] [32] |
| BioGRID Tab 3.0 | .tab3.zip | 167-172 MB | Script-based analysis, custom pipelines [29] [32] |
| PSI MITAB 2.5 | .mitab.zip | 169-176 MB | Standardized tabular analysis [29] [32] |
| Organism-Specific | Varies | 61-188 MB | Species-focused research [29] |
| Chemical Data | .chemtab.zip | ~1.3 MB | Chemical biology, drug discovery [29] |
| Post-Translational Modifications | .ptm.zip | ~56 MB | Signaling pathway analysis [29] |
Legacy Format Considerations
BioGRID maintains several legacy formats including BioGRID TAB 2.0, TAB 1.0, and PSI-MI XML 1.0 to ensure backward compatibility with existing research pipelines [32]. However, for new projects, the use of current recommended formats is strongly advised as they contain the most up-to-date data structure improvements and comprehensive interaction records. The legacy formats are primarily recommended only for maintaining compatibility with existing legacy projects [32].
IntAct Data Access Framework
Data Model and Access Capabilities
IntAct employs a sophisticated data model that supports two levels of curation detail: full IMEx-level annotation and MIMIx-compatible entries [31]. This flexible framework allows researchers to access data at different levels of granularity based on their specific requirements. The database provides both web-based query interfaces and programmatic access options, enabling interactive exploration and large-scale computational analysis. IntAct's website has been specifically redesigned to enhance user experience, featuring improved search processes and more detailed graphical displays of interaction results [31].
Data Export and Integration
IntAct supports multiple data export formats that facilitate various analytical approaches. The resource provides specialized data visualization tools that allow researchers to generate interaction network diagrams directly from query results. Additionally, IntAct data is available in formats compatible with the Semantic Web, enhancing computational accessibility and integration with other linked data resources [31]. This commitment to standardized data representation ensures that IntAct datasets can be seamlessly incorporated into broader bioinformatics workflows and analytical pipelines.
Experimental and Computational Methodologies
Practical Workflow for PPI Data Retrieval
The following workflow diagram illustrates a standardized protocol for accessing PPI data from major databases:
Diagram 1: PPI Data Retrieval Workflow
Protocol 1: Targeted Gene Query via Web Interface
Purpose: To extract interaction data for specific candidate genes through graphical web interfaces.
Materials:
- Computer with internet access
- Supported web browser (Chrome, Firefox, or Edge)
- List of target gene identifiers
Procedure:
- Navigate to the BioGRID or IntAct website using a supported web browser [29] [31].
- Locate the search interface, typically prominently displayed on the homepage.
- Input official gene symbols or identifiers for your target genes.
- Apply relevant filters to restrict results by:
- Organism (e.g., Homo sapiens)
- Experimental system (e.g., physical vs. genetic interactions)
- Interaction detection method
- Review the returned interaction network visually.
- Select appropriate download format based on intended use (refer to Table 2).
- Export the data file to your local analysis environment.
Technical Notes: For BioGRID, the "Multi-Validated" dataset filter can be applied to obtain high-confidence physical interactions [32]. For IntAct, leverage the fine-grained annotation to filter interactions by specific experimental evidence.
Protocol 2: Bulk Data Download for Network Analysis
Purpose: To download complete datasets for comprehensive network analysis or integration with internal data.
Materials:
- Stable internet connection with sufficient bandwidth
- Adequate local storage capacity (250MB+ recommended)
- Data extraction tool (scripting environment or archive utility)
Procedure:
- Access the BioGRID download repository at https://downloads.thebiogrid.org/ [29] [30].
- Identify the most current release directory (e.g., Release 5.0.251) [29].
- Select the appropriate file format based on analytical requirements:
- Use PSI25 for computational integration
- Use TAB3 for custom script-based analysis
- Use MITAB for standardized tabular processing [32]
- Download the compressed data file.
- Extract the archive using appropriate decompression tools.
- Validate data integrity through record counts or checksum verification.
Technical Notes: For large-scale analyses, consider using BioGRID's REST service with JSON formatting for efficient programmatic access [32]. Always use the most recent release for new projects to ensure data comprehensiveness [29].
Protocol 3: Cross-Database Integration Methodology
Purpose: To integrate complementary PPI data from multiple databases for comprehensive coverage.
Materials:
- Data from at least two PPI databases (e.g., BioGRID and STRING)
- Data integration pipeline (custom scripts or workflow tools)
- Identifier mapping resources
Procedure:
- Download datasets from selected databases using appropriate formats.
- Standardize protein identifiers across datasets using mapping resources.
- Apply confidence filters specific to each database's metrics.
- Merge interaction records while preserving source attribution.
- Resolve conflicting interactions through evidence weighting.
- Generate a unified interaction network for analysis.
Technical Notes: Systematic comparisons indicate that combined use of STRING and UniHI covers approximately 84% of experimentally verified PPIs, while adding IID and hPRINT extends coverage to 94% of total available interactions [16]. BioGRID contributes significantly to experimentally verified interactions, with STRING providing approximately 71% of exclusive experimentally verified hits [16].
Table 3: Essential Research Reagents and Computational Resources for PPI Research
| Resource Type | Specific Tool/Reagent | Function/Application |
|---|---|---|
| Core Databases | BioGRID [29] [22] | Comprehensive curated protein, genetic and chemical interactions |
| IntAct [31] | Fine-grained molecular interaction data with deep annotation | |
| STRING [28] [16] | Known and predicted protein-protein interactions with confidence metrics | |
| Specialized Resources | BioGRID-ORCS [22] | CRISPR screening data and results |
| BioGRID Themed Projects [29] [22] | Disease-focused interaction sets (Alzheimer's, COVID-19, etc.) | |
| Analytical Formats | PSI-MI XML 2.5 [32] | Standardized format for data exchange and computational analysis |
| BioGRID TAB 3.0 [32] | Tab-delimited format for custom analytical pipelines | |
| Software & Libraries | Osprey Network Visualization [32] | Network visualization and analysis of interaction data |
| Graph Neural Networks [28] | Deep learning approaches for PPI prediction and analysis |
Advanced Data Integration and Analysis Techniques
Computational Framework for PPI Network Analysis
The following diagram illustrates a sophisticated computational pipeline for integrated PPI data analysis:
Diagram 2: Computational Analysis Pipeline
Deep Learning Architectures for PPI Analysis
Modern PPI research increasingly incorporates deep learning frameworks to extract meaningful patterns from complex interaction data. Several architectural approaches have demonstrated particular utility:
Graph Neural Networks (GNNs): These networks directly operate on graph-structured data, making them ideally suited for PPI networks. Variants such as Graph Convolutional Networks (GCNs) aggregate information from neighboring nodes to capture local patterns, while Graph Attention Networks (GATs) introduce attention mechanisms that adaptively weight the importance of different interactions [28].
Multi-Modal Frameworks: Advanced systems like the AG-GATCN framework integrate multiple architectural components (GAT and Temporal Convolutional Networks) to enhance robustness against biological noise in PPI data [28].
Representation Learning Methods: Architectures such as the Deep Graph Auto-Encoder (DGAE) combine canonical auto-encoders with graph auto-encoding mechanisms to enable hierarchical representation learning for PPI characterization [28].
These computational approaches are particularly valuable for addressing inherent challenges in PPI data analysis, including data imbalances, biological variations, and high-dimensional feature sparsity [28].
Effective access to PPI data through appropriate download formats and web interfaces is a critical competency for modern biological research. BioGRID and IntAct provide complementary resources with distinct strengths—BioGRID offers extensive curation volume and specialized datasets, while IntAct provides granular experimental annotation. The selection of specific data formats should be guided by analytical objectives, with PSI-MI XML 2.5 and BioGRID TAB 3.0 representing optimal choices for most new research initiatives. As the field advances, integration of multiple data sources and application of sophisticated computational methods like graph neural networks will increasingly drive discoveries in systems biology and drug development. Researchers are encouraged to leverage the standardized protocols and resource comparisons presented in this guide to optimize their PPI data access strategies, ensuring robust and reproducible research outcomes in the evolving landscape of interaction bioinformatics.
From Data to Biological Insight: Strategies for Integration and Specialized Network Construction
Protein-protein interaction (PPI) networks are fundamental to systems biology, providing a framework for understanding cellular machinery, signal transduction, and disease mechanisms [33]. The set of all interactions within an organism forms a protein interaction network (PIN), which serves as a critical tool for studying cellular behavior [34]. While public databases such as IntAct, BioGRID, and STRING provide vast repositories of interaction data, simply taking the union of data from these sources constitutes a naive approach that fails to address critical challenges including identifier inconsistencies, varying evidence types, and confidence scoring disparities [35] [22]. A robust integrated network requires sophisticated methodologies that move beyond simple data aggregation to create biologically coherent and analytically reliable networks suitable for hypothesis generation and validation in biomedical research.
The process of building these networks must address multiple dimensions of complexity. First, PPI data originates from diverse experimental techniques (e.g., yeast two-hybrid, mass spectrometry) and computational predictions, each with different reliability metrics and systematic biases [33] [35]. Second, the heterogeneity of nodes (proteins) and edges (interactions) requires semantic integration of biological annotations from ontologies like Gene Ontology (GO) and pathway databases such as KEGG and Reactome [33] [35]. Finally, effective visualization and analysis demand specialized software platforms that can handle the scale and complexity of integrated networks while providing analytical capabilities for biological discovery [36] [34]. This guide provides a comprehensive technical framework for constructing robust integrated PPI networks, with specific protocols and resources for research scientists and drug development professionals.
Core PPI Databases and Their Characteristics
A strategic integration approach begins with understanding the specialized strengths and limitations of available databases. The table below summarizes major PPI resources and their distinctive properties.
Table 1: Key Protein-Protein Interaction Databases and Resources
| Database Name | Primary Focus | Evidence Types | Update Frequency | Key Features |
|---|---|---|---|---|
| BioGRID [22] | Physical & genetic interactions | Curated from literature, high- & low-throughput experiments | Monthly | Extensive curation with >2.2 million non-redundant interactions; themed curation projects for specific diseases |
| STRING [35] | Functional & physical associations | Experimental, predictive, co-expression, text mining | Regularly updated | Comprehensive confidence scoring; cross-species transfer via interologs; regulatory networks |
| IntAct [35] | Molecular interaction data | Curated experiments from literature | Regular updates | IMEx consortium member; standardized data formats |
| MINT [35] | Experimentally verified PPIs | Focus on high-throughput experiments | Regular updates | Specialized in molecular interactions |
| HPRD [28] | Human protein reference | Manual curation from literature | Not specified | Human-specific data with enzymatic and localization data |
| DIP [28] | Experimentally verified PPIs | Curated experiments | Not specified | Database of Interacting Proteins |
| Reactome [35] | Pathway-centered interactions | Expert-curated pathways | Regular updates | Hierarchically nested pathway modules; pathway enrichment analysis |
Understanding the scale and composition of PPI data is essential for designing integration strategies. The following table provides comparative metrics for major resources (based on latest available data).
Table 2: Comparative Quantitative Metrics of PPI Resources
| Database | Publications | Interactions | Organisms | Confidence Scoring | Specialized Networks |
|---|---|---|---|---|---|
| BioGRID [22] | 87,393+ | >2.25M non-redundant | Multiple | Based on experimental evidence type | Themed projects (Autism, Alzheimer's, COVID-19) |
| STRING [35] | Not specified | Comprehensive coverage | 1000s of organisms | Probability score (0-1) for each association | Physical, regulatory, and functional networks |
| CORUM [28] | Not specified | Focus on complexes | Human | Experimental validation | Protein complexes specifically |
Methodological Framework: From Simple Union to Robust Integration
Core Challenges in PPI Network Integration
Building a robust integrated PPI network requires addressing several fundamental challenges that extend beyond simple data aggregation. The high number of nodes and connections in real PINs demands significant computational resources and can complicate graphical rendering and analysis [34]. Furthermore, the heterogeneity of nodes (proteins) and edges (interactions) creates integration complexity, particularly when combining data from multiple sources with different identifier systems and annotation standards [33]. The ability to annotate proteins and interactions with biological information extracted from ontologies (e.g., Gene Ontology) enriches PINs with semantic information but substantially complicates their visualization and analysis [33] [34]. Additionally, the availability of numerous data formats for representing PPI and PINs data creates interoperability challenges that must be addressed through standardized conversion pipelines [34].
Strategic Integration Workflow
The following diagram illustrates a comprehensive workflow for robust PPI network integration, moving systematically from data acquisition to functional validation:
Diagram 1: PPI Network Integration Workflow (width=760px)
Advanced Integration Protocols
Protocol 1: Identifier Mapping and Standardization
Effective integration requires resolving identifier inconsistencies across databases. This protocol ensures uniform protein identification:
- Source Data Acquisition: Download PPI data from multiple sources (BioGRID, STRING, IntAct) in standard formats (PSI-MI, TSV, or XML).
- Identifier Extraction: Extract all protein identifiers, noting the database of origin and identifier type (UniProt, Ensembl, Entrez Gene, RefSeq).
- Mapping Service Utilization: Use robust mapping services (UniProt ID Mapping, BioMart, g:Profiler) to convert all identifiers to a standardized namespace (recommended: UniProt KB accession numbers).
- Ambiguity Resolution: Manually resolve identifier ambiguities through sequence-based matching when automatic methods fail.
- Identifier Consolidation: Create a master mapping table that preserves all original identifiers while maintaining the standardized identifier as the primary key.
Protocol 2: Evidence-Weighted Confidence Scoring
Simple union approaches treat all interactions equally, regardless of evidence quality. This advanced protocol implements evidence-weighted confidence assessment:
Evidence Channel Classification: Categorize interaction evidence into distinct channels:
- Experimental (high-throughput vs. low-throughput)
- Computational predictions (genomic context, sequence-based)
- Database annotations (curated pathways)
- Text mining co-occurrence
Channel-Specific Scoring: Calculate confidence scores for each evidence channel using platform-specific metrics (e.g., STRING's neighborhood, fusion, and co-occurrence scores) [35].
Probabilistic Integration: Combine channel-specific scores using probabilistic integration, assuming evidence independence across channels. The combined confidence score is computed as: P(combined) = 1 - Π(1 - P_i) for i evidence channels
Threshold Application: Apply organism- and context-specific confidence thresholds (typically 0.7-0.9 for high-confidence networks).
Directionality Annotation: For regulatory networks, incorporate directionality information using natural language processing of literature and curated pathway databases [35].
Protocol 3: Semantic Integration of Functional Annotations
Moving beyond structural networks to functionally annotated networks enables deeper biological insights:
Ontology Resource Identification: Identify relevant ontologies (Gene Ontology, KEGG pathways, Reactome pathways) for functional annotation.
Annotation Mapping: Map standardized protein identifiers to functional annotations using services provided by EBI QuickGO, KEGG API, or custom mapping pipelines.
Enrichment Analysis Preparation: Precompute background gene sets appropriate for your organism and research context.
Semantic Similarity Calculation: Implement semantic similarity measures (Resnik, Lin, or Wang methods) to quantify functional relationships between proteins beyond direct interactions.
Annotation Integration: Integrate functional annotations as node attributes in the network for subsequent visualization and analysis.
Computational Implementation and Visualization Solutions
Software Platforms for PPI Network Analysis
Multiple software platforms support PPI network visualization and analysis, each with distinctive capabilities. The following table compares key tools used in robust network integration.
Table 3: Software Platforms for PPI Network Visualization and Analysis
| Software Tool | License Model | Key Features | Integration Capabilities | Analysis Functions |
|---|---|---|---|---|
| Cytoscape [36] [34] | Open source | Extensible via apps, multiple layout algorithms | Import from multiple formats, REST API | Network analysis, clustering, functional enrichment |
| NAViGaTOR [34] | Closed source | High-performance 2D/3D visualization | GraphML and other standard formats | Specialized for large network visualization |
| PINA [37] | Web platform | Integrated analysis of six databases | Built-in multi-database integration | Network construction, filtering, analysis |
Advanced Layout Algorithms for Network Visualization
Effective visualization requires appropriate layout algorithms that highlight biologically meaningful patterns. The following diagram illustrates the algorithmic decision process for selecting optimal visualization strategies:
Diagram 2: Layout Algorithm Selection Guide (width=760px)
Building robust integrated PPI networks requires both computational tools and biological resources. The following table details essential components of the network analysis toolkit.
Table 4: Essential Research Reagents and Resources for PPI Network Analysis
| Resource Category | Specific Tools/Resources | Function/Purpose | Implementation Considerations |
|---|---|---|---|
| Data Retrieval Tools | STRING API [35], BioGRID web services [22], IntAct PSICQUIC | Programmatic access to PPI data | Rate limiting, format conversion, caching strategies |
| Identifier Mapping | UniProt ID Mapping, BioMart, g:Profiler | Standardizing protein identifiers across databases | Batch processing for large datasets, ambiguity resolution |
| Analysis Environments | Cytoscape [36], R/Bioconductor, Python NetworkX | Network construction, analysis, and visualization | Plugin architecture (Cytoscape), library dependencies |
| Specialized Algorithms | MCL clustering, NetworkAnalyzer, CentiScaPe [36] | Identification of functional modules and key proteins | Parameter optimization for biological networks |
| Validation Resources | CRISPR screening data (BioGRID ORCS) [22], Gene Ontology annotations | Biological validation of network predictions | Statistical frameworks for enrichment analysis |
Validation Framework and Biomedical Applications
Multi-Level Validation Strategy
Robust network integration requires systematic validation across multiple biological scales:
Topological Validation: Assess network properties using graph metrics (degree distribution, clustering coefficient, betweenness centrality) to ensure biological plausibility.
Functional Validation: Perform enrichment analysis using Gene Ontology, KEGG pathways, and disease annotations to verify that integrated networks recover known biological relationships.
Experimental Validation: Design wet-lab experiments (co-immunoprecipitation, FRET, yeast two-hybrid) to test high-confidence novel predictions from the integrated network.
Cross-Species Validation: Leverage interolog mapping [35] to transfer interactions across species and validate conserved modules.
Case Study: Integrating TGFβ and LKB1 Signaling Pathways
The PINA platform demonstrated the power of integrated network analysis by revealing previously unknown connections between LKB1 and TGFβ signaling pathways [37]. This analysis:
Integrated Data Sources: Combined PPI data from six primary databases using identifier standardization and confidence scoring.
Applied Advanced Algorithms: Utilized network clustering and topological analysis to identify bridging components.
Generated Testable Hypotheses: Proposed specific molecular mechanisms for crosstalk between these critical signaling pathways.
Identified Novel Interactions: Revealed potential competitive interactors of p53 and c-Jun that could not be detected in single-database analyses.
This case study exemplifies how robust integration moves beyond simple unions to create novel biological insights with potential therapeutic implications.
Emerging Trends and Future Directions
The field of PPI network integration is rapidly evolving, with several emerging trends shaping future methodologies. Deep learning approaches, particularly graph neural networks (GNNs), are revolutionizing PPI prediction and analysis [28]. Architectures such as Graph Convolutional Networks (GCNs), Graph Attention Networks (GATs), and Graph Autoencoders can capture complex patterns in network data that traditional methods miss [28]. These approaches enable automatic feature learning from protein sequences, structures, and existing network topology, potentially overcoming limitations of manually engineered features.
Another significant trend is the move toward more specific interaction typing in composite databases. STRING's introduction of distinct physical and regulatory networks represents a major advancement in interaction specificity [35]. This enables researchers to move beyond undirected functional associations to analyze directed regulatory relationships and physical binding events separately, providing more mechanistic insights. The application of fine-tuned language models to extract directional information from literature further enhances these capabilities [35].
From a technological perspective, the tension between open, extensible platforms like Cytoscape and high-performance, often closed-source tools like NAViGaTOR continues to drive innovation [34]. Open-source tools benefit from large developer and user communities that ensure long-term sustainability and continuous feature expansion through plugins and extensions. Conversely, specialized closed-source tools can optimize performance for specific use cases, such as visualizing extremely large networks with thousands of nodes [34]. The emerging solution of hybrid architectures, where core visualization engines handle performance-critical tasks while plugin systems accommodate analytical extensions, may offer the best of both approaches.
Future developments will likely focus on dynamic network modeling, integration of single-cell omics data, and application of large language models for knowledge extraction from literature. These advancements will further enhance our ability to build biologically accurate, context-specific PPI networks that move far beyond simple database unions to become predictive models of cellular behavior.
Protein-protein interactions (PPIs) are fundamental to nearly every biological process, and PPI networks provide critical insight into the underlying mechanisms of disease and cellular function. Researchers rarely rely on a single data source; instead, they often turn to multiple public PPI databases such as BioGRID, IntAct, HPRD, and MINT to construct comprehensive networks. However, this practice introduces a significant methodological challenge: how to effectively integrate these disparate datasets to build a robust and biologically relevant PPI network without incurring high false positive rates. The k-votes integration method was developed specifically to address this challenge, providing a systematic, evidence-based approach for combining multiple PPI databases. This method moves beyond the traditional simple union of datasets by requiring that interactions are supported by multiple independent sources, thereby substantially improving the reliability of the resulting integrated network for downstream analysis in biomedical research and drug development.
Understanding the k-votes Methodology
Core Conceptual Framework
The k-votes method operates on a committee-based voting principle. When integrating multiple PPI databases, each database is treated as an independent "committee member" that casts a vote on whether a specific protein-protein interaction exists. The core premise is that an interaction is included in the final integrated network only if it receives a consensus of at least k votes from the committee of source databases.
Formally, given n PPI database networks represented as G₁, G₂, G₃, ..., Gₙ, where each Gᵢ = <Vᵢ, Eᵢ> consists of a set of vertices (proteins) Vᵢ and edges (interactions) Eᵢ, the integrated network Ĝ using the k-votes method is mathematically defined as:
Ĝ = {Gᵢ₁ ∪ Gᵢ₂ ∪ Gᵢ₃ ∪ ... ∪ Gᵢₖ} for all subsets {Gᵢ₁, Gᵢ₂, Gᵢ₃, ..., Gᵢₖ} of {G₁, G₂, G₃, ..., Gₙ} [38].
This means an edge (interaction) is included in Ĝ if and only if it appears in at least k of the n source databases. The value of k can be tuned from 1 to n, with higher values requiring more substantial evidence for an interaction's inclusion.
Contrasting Traditional Union with k-votes Approach
The traditional approach to integrating multiple PPI databases has been the union method (k=1), where any interaction found in any single database is included in the integrated network. While this maximizes coverage, it comes at the cost of potential false positives due to database-specific errors, different curation standards, or identifier mapping issues [24]. The k-votes method introduces a quality filter by requiring multi-database support, effectively trading off some sensitivity for greatly improved specificity.
Table 1: Comparison of Integration Approaches for PPI Networks
| Integration Method | Description | Advantages | Disadvantages |
|---|---|---|---|
| Union (k=1) | Includes interactions present in any single database | Maximum sensitivity, comprehensive coverage | Higher false positive rate, includes database-specific errors |
| k-votes (k≥2) | Requires interactions to be present in at least k databases | Higher specificity, reduced false positives, more reliable interactions | Potentially misses some true interactions (lower sensitivity) |
| Intersection (k=n) | Requires interactions to be present in all databases | Maximum specificity, highest confidence interactions | Very low sensitivity, excludes many true interactions |
Experimental Validation and Performance Analysis
Original Study Design and Database Selection
The k-votes method was systematically evaluated in a landmark study that integrated seven major public PPI databases: BioGRID, DIP, HPRD, IntAct, MINT, REACTOME, and SPIKE [38]. This created a comprehensive framework for assessing the method's performance across different consensus thresholds. The study focused exclusively on Homo sapiens PPIs to ensure biological relevance and consistency, with all protein identifiers unified using Entrez IDs to resolve database-specific nomenclature issues.
Table 2: PPI Databases Used in the Original k-votes Validation Study
| Database | Number of Proteins | Number of Interactions | Primary Focus and Characteristics |
|---|---|---|---|
| BioGRID | 8,204 | 33,625 | Publication-based curation of protein and genetic interactions across major model organisms [38] |
| DIP | 1,137 | 1,509 | Experimentally determined interactions with computational correction and expert review [38] |
| HPRD | 9,553 | 38,802 | Comprehensive human protein database with interactions, modifications, and disease associations [38] |
| IntAct | 7,495 | 30,965 | Molecular interactions from literature or direct curator submissions using comprehensive annotation [38] |
| MINT | 5,230 | 15,353 | Experimentally verified PPIs mined from scientific literature by expert curators [38] |
| REACTOME | 3,599 | 74,490 | Manually curated and peer-reviewed pathway database with interactions from pathway perspectives [38] |
| SPIKE | 6,927 | 23,224 | Thoroughly curated database of human signaling pathways [38] |
Evaluation Framework and Metrics
To determine the optimal value of k, the study employed a rigorous evaluation framework using the Structural Clustering Algorithm for Networks (SCAN). This algorithm identifies functional modules (clusters) in networks based on the structural similarity of connected vertices, calculated using common neighbors [38]. The quality of the integrated networks generated with different k values was assessed using multiple statistical and biological measures:
- Modularity (QN): Measures the quality of network clustering by comparing the density of edges within clusters to the expected density if edges were distributed randomly [38].
- Similarity-Based Modularity (QS): Addresses the resolution limit problem of standard modularity by incorporating structural similarity metrics, particularly important for networks with strongly varying cluster sizes [38].
- Clustering Score: Evaluates the biological coherence of identified modules.
- Enrichment Analysis: Assesses the functional relevance of identified modules using gene ontology and pathway analysis.
Key Findings and Optimal k Determination
The study demonstrated that the k-votes method significantly outperformed the traditional union approach in both statistical significance and biological meaning. Through comprehensive analysis across all seven k values (1-7), researchers determined that k=2 provided the optimal balance between network coverage and reliability [38]. The integrated network at k=2, composed of interactions confirmed in at least two PPI databases, showed superior functional module quality compared to both the union approach (k=1) and higher consensus thresholds (k>2). This finding indicates that requiring interactions to have just one additional independent source of evidence substantially reduces false positives while maintaining sufficient coverage of the true interactome.
Practical Implementation Protocol
Data Acquisition and Preprocessing
Implementing the k-votes method begins with acquiring current data from multiple PPI databases. As of 2025, BioGRID alone contains over 2.2 million non-redundant interactions from more than 87,000 publications [22], highlighting the massive scale of modern PPI data. The implementation protocol involves these critical steps:
- Download PPI data from your selected databases (BioGRID, IntAct, MINT, etc.) in standardized formats such as PSI-MI TAB or via API access when available.
- Filter for organism-specific data (e.g., Homo sapiens) to ensure biological consistency.
- Map protein identifiers to a consistent namespace (e.g., Entrez Gene IDs, UniProt accessions) across all databases using mapping tables or services.
- Resolve duplicates within individual databases to ensure each unique protein-protein pair is represented only once per database.
- Store processed data in a structured format that maintains the source database attribution for each interaction.
Integration Workflow Using k-votes
The core integration process follows a systematic workflow that can be implemented using bioinformatics scripting tools such as Python or R:
Diagram 1: k-votes Integration Workflow
Computational Tools and Implementation
The k-votes method can be implemented using various computational approaches:
Custom Script Implementation:
Available Resources and Tools: While the original k-votes method was implemented using custom scripts, current PPI meta-databases such as APID (Agile Protein Interaction DataAnalyzer) offer pre-integrated interaction data from multiple sources, though they may not explicitly implement the k-votes framework [39]. Researchers can also leverage workflow platforms like Galaxy or Cytoscape with appropriate plugins to create customized integration pipelines.
Table 3: Key Research Resources for PPI Network Integration Studies
| Resource Category | Specific Examples | Function and Application |
|---|---|---|
| Primary PPI Databases | BioGRID, IntAct, MINT, HPRD | Source databases providing experimentally validated protein-protein interactions from literature curation [22] [24] |
| Pathway Databases | REACTOME, SPIKE | Contextualize PPIs within broader signaling pathways and biological processes [38] |
| Standardized Formats | PSI-MI (Proteomics Standards Initiative - Molecular Interaction) | Enable consistent data exchange and integration across different databases and tools [24] |
| Identifier Mapping Services | UniProt ID Mapping, BioMart | Resolve different protein identifiers across databases to a consistent namespace [38] |
| Network Analysis Platforms | Cytoscape with dedicated plugins | Visualize, analyze, and integrate PPI networks with additional omics data layers |
| Clustering Algorithms | SCAN (Structural Clustering Algorithm for Networks) | Identify functional modules in integrated PPI networks based on structural similarity [38] |
Application in Biomedical Research and Drug Development
The k-votes integration method has significant implications for biomedical research and pharmaceutical development. By producing more reliable PPI networks, it enhances the identification of disease-relevant protein modules and druggable targets. The approach aligns with the recognition that disease-associated genes often encode proteins that interact with each other [38]. For drug development professionals, the method reduces the risk of pursuing false leads based on single-database interactions while highlighting high-confidence targets supported by multiple independent sources.
The k-votes framework has also inspired similar voting-based integration approaches in other domains, including causal network inference [40] and crowdsourcing systems [41], demonstrating its versatility as a data integration paradigm. These applications share the core principle that requiring consensus from multiple independent sources significantly improves result reliability.
Advanced Considerations and Future Directions
Weighted Voting Extensions
While the standard k-votes method treats all databases equally, advanced implementations can incorporate weighted voting based on database quality metrics or specific research contexts. For example, databases with more rigorous curation standards or experimental validation could be assigned higher voting weights. Similarly, context-specific weighting could prioritize databases with stronger coverage of particular biological domains, such as signaling pathways or disease-associated proteins.
Temporal and Contextual Integration
Modern PPI data increasingly includes contextual information such as tissue specificity, post-translational modifications, and temporal dynamics. The k-votes framework can be extended to incorporate these dimensions by implementing context-aware voting schemes that only require consensus within specific biological contexts. BioGRID's ongoing curation efforts now include over 560,000 non-redundant post-translational modification sites [22], highlighting the growing importance of these contextual data layers.
Integration with Complementary Data Types
Future enhancements to the k-votes approach could integrate PPI data with complementary functional genomics data, such as genetic interaction networks (also available in BioGRID), gene co-expression patterns, and phylogenetic profiles. This multi-dimensional integration would create more comprehensive cellular network models while maintaining the core voting principle for reliability improvement.
Diagram 2: Multi-dimensional k-votes Integration
Constructing Tissue and Tumor-Specific PPI Networks with Tools like SPECTRA
Protein-protein interaction (PPI) networks have become fundamental to understanding cellular functions, yet traditional PPI repositories present a significant limitation: they typically represent aggregate interactions across all cellular contexts, ignoring the specific tissues or pathological states where these interactions actually occur [42]. This oversight is particularly problematic for understanding human disease, as proteins can form tissue-selective complexes while remaining inactive in other tissues, and many diseases manifest specifically in certain tissues [42] [43]. The integration of protein interaction data with tissue-specific expression information has given rise to Tissue-Specific PPI (TS-PPI) networks - subgraphs of global PPI networks where both interacting proteins are expressed in selected tissues [42].
This technical guide examines the construction and analysis of TS-PPI networks, with particular focus on the SPECTRA framework, and places these resources within the broader ecosystem of PPI data tools including IntAct and BioGRID. For researchers in drug development and systems biology, these tools provide critical insights into context-specific protein function and enable identification of disease-specific therapeutic targets that might be obscured in global interactome maps.
Foundations of Tissue-Specific Network Construction
Core Data Components and Integration Methodology
Constructing biologically meaningful TS-PPI networks requires the integration of two primary data types: protein interaction data and tissue-specific expression information.
Protein-Protein Interaction Data is sourced from major repositories that collectively document hundreds of thousands of experimentally determined interactions:
- BioGRID: Contains over 2.2 million non-redundant interactions curated from more than 87,000 publications [22] [44]
- IntAct: Provides both data and tools for textual and graphical representation of protein interactions [42]
- MINT, DIP, and HPRD: Additional primary sources contributing to the overall interaction coverage [42]
Integrative databases like STRING combine physical interaction data with predicted interactions from text mining and genomic features, while IRefIndex and ConsensusPathDB provide unified access to interactions from multiple sources [42].
Expression Data is obtained from authoritative repositories profiling normal and pathological tissues:
- The Cancer Genome Atlas (TCGA): Collects complete high-throughput genome data for specific cancer tissues [42]
- Human Protein Atlas (HPA): Contains histological images, transcription expression levels, protein expression profiles, and subcellular localization data [42] [45]
- ArrayExpress and GEO: Include gene expression data from microarray and high-throughput sequencing experiments [42]
Emerging Methods for Association Prediction
While expression-based filtering remains a common approach, recent advances leverage protein co-abundance across thousands of proteomic samples to predict functional associations. This method outperforms both mRNA coexpression and protein cofractionation in recovering known complex members (AUC = 0.80 ± 0.01 for coabundance vs. 0.69 ± 0.01 for cofractionation and 0.70 ± 0.01 for mRNA coexpression) [45]. This suggests post-transcriptional processes drive most of the predictive power for protein associations rather than regulation of gene expression alone [45].
Table 1: Data Sources for Constructing Tissue-Specific PPI Networks
| Data Type | Primary Sources | Key Features | Coverage |
|---|---|---|---|
| Protein Interactions | BioGRID, IntAct, MINT, HPRD, DIP | Experimentally validated physical interactions | >2.2M non-redundant interactions (BioGRID) [22] |
| Integrated PPI Databases | STRING, IRefIndex, ConsensusPathDB | Unified access, confidence scoring, functional annotations | Combines multiple primary sources [42] |
| Expression Data | Human Protein Atlas, TCGA, ArrayExpress, GEO | Tissue/tumor sequencing, protein abundance, histological images | 7,811+ proteomic samples across 11 tissues [45] |
| Protein Complex Reference | CORUM | Curated database of protein complexes | Ground truth for validation [45] |
SPECTRA: A Framework for TS-PPI Network Analysis
Architecture and Data Integration
SPECTRA (SPECific Tissue/Tumor Related PPI networks Analyzer) is a comprehensive knowledge base designed specifically for building and comparing tissue or tumor-specific PPI networks [42]. Its architecture integrates 16,435 protein-coding genes and 175,841 gene interactions with 1,350,637 tissue-specific gene expression data entries covering 107 normal tissues and 2,171,808 tumor-specific expression data entries spanning 160 different tumors [46].
The framework applies a rigorous integration methodology where each gene-tissue pair is assigned a unique positive expression score derived from the average normalized expression value of the gene in that tissue across different datasets [46]. Expression scores in SPECTRA range from 3.566 to 17.366 for tissues and from 0.01 to 17.343 for tumors, providing a quantitative basis for filtering interactions [46].
Workflow and User Interface
SPECTRA implements a structured workflow for TS-PPI network construction:
- Gene Selection: Users input a set of genes or proteins of interest
- Tissue/Tumor Selection: Specify normal tissues, tumors, or both for analysis
- Expression Data Selection: Choose from integrated datasets (Protein Atlas, ArrayExpress, TCGA)
- Interaction Data Selection: Select PPI sources (BioGRID, HPRD, MIPS, IntAct)
- Threshold Application: Set minimum expression values for gene inclusion [46]
The system generates TS-PPI networks where interactions are retained only if both participating proteins demonstrate expression above threshold in the selected tissue context. SPECTRA provides both visualization capabilities through Cytoscape integration and analytical functions for comparing networks across different tissues or states [42] [46].
Figure 1: SPECTRA Workflow for TS-PPI Network Construction and Analysis
Analytical Methods for TS-PPI Networks
Topological Analysis of Networks
Once constructed, TS-PPI networks require specialized analytical approaches to extract biologically meaningful insights. Topological analysis identifies strategically important proteins through several key metrics:
- Degree Centrality: Number of connections a node has; proteins with high degree ("hubs") are often crucial for network integrity [47] [48]
- Betweenness Centrality: Measures how often a node acts as a bridge along shortest paths; nodes with high betweenness ("bottlenecks") have more control over information flow [47] [48]
- Closeness Centrality: Indicates how close a node is to all others; higher values suggest more central positioning in the network [48]
In practice, researchers often identify proteins with top 10% highest degree or betweenness centrality as the "backbone" of the network for further investigation [47]. For example, in a study of Heroin Use Disorder, JUN possessed the largest degree while PCK1 showed the highest betweenness centrality, suggesting their central roles in the associated PPI network [47].
Differential Network Alignment
SPECTRA implements specialized algorithms for comparing TS-PPI networks across different conditions. The GASOLINE algorithm addresses the local differential alignment problem, identifying conserved sub-regions that maximize expression differences between aligned genes [46].
The modified GASOLINE algorithm for expression-weighted comparison involves:
- Bootstrap Phase: Identification of orthologous proteins across networks as seeds
- Iterative Phase: Repeated addition or removal of nodes to maximize alignment score
- Scoring: Incorporation of expression differences through LogFold change calculations
- Post-processing: Ranking alignments by Index of Structural Conservation (ISC) score [46]
This approach enables researchers to identify network regions with significant expression differences between normal and pathological states, or across different tissues, highlighting potential mechanistic differences.
Figure 2: Differential Network Alignment Process
Alternative Tools and Databases
While SPECTRA provides comprehensive functionality, several alternative tools offer complementary approaches to tissue-specific network analysis:
TissueNet v.2 offers both qualitative and quantitative views of query proteins and their PPIs across tissues, highlighting tissue-specific and globally-expressed proteins [43]. It incorporates RNA-sequencing data from GTEx and HPA, protein expression profiles from antibody staining, and supports differential expression analysis [43]. A key feature is user-defined expression thresholds for tissue associations via an interactive sliding bar [43].
APPIC (Atlas of Protein-Protein Interactions in Cancer) focuses specifically on cancer subtypes, identifying PPI networks shared by cohorts of patients across 10 cancer types and 26 subtypes [49]. It integrates biological and clinical information from HPA, HGNC, g:Profiler, cBioPortal, and Clue.io, supporting both 2D and 3D network visualizations [49].
Co-abundance Association Atlas represents a recent methodology that leverages protein co-abundance across 7,811 human biopsies to score association likelihood for 116 million protein pairs across 11 human tissues [45]. This approach demonstrates that over 25% of associations are tissue-specific, with less than 7% attributable solely to differences in gene expression [45].
Table 2: Comparison of Tissue-Specific PPI Tools and Databases
| Tool | Primary Focus | Key Features | Data Sources | Use Case |
|---|---|---|---|---|
| SPECTRA | General tissue & tumor networks | Differential alignment, multi-network comparison | BioGRID, HPRD, MIPS, IntAct, Protein Atlas, TCGA, ArrayExpress, GEO [42] [46] | Comparing networks across multiple tissues or states |
| TissueNet v.2 | Human tissue interactomes | User-defined expression thresholds, differential expression view | GTEx, HPA, BioGRID, IntAct, MINT, DIP [43] | Exploring tissue-specificity of query proteins |
| APPIC | Cancer subtype networks | Patient clustering, therapeutic target identification | STRING, cBioPortal, HPA, Clue.io [49] | Identifying subtype-specific mechanisms in cancer |
| Co-abundance Atlas | Protein association prediction | Co-abundance scoring across biopsies | Proteomic samples from 50 studies [45] | Discovering tissue-specific functional associations |
Experimental Validation Methods
Computational predictions of TS-PPIs require experimental validation, with several methods commonly employed:
- Cofractionation-MS: Protein complexes are separated by chromatography and subsequent fractions are analyzed by mass spectrometry to identify co-eluting proteins [45]
- Affinity Purification-MS: Bait proteins are purified with binding partners and identified via mass spectrometry [45]
- Yeast Two-Hybrid: Detects binary interactions through reconstitution of transcription factors [50]
- Protein-fragment Complementation: Reconstituition of protein function when fragments brought together by interacting proteins [45]
Recent frameworks for brain tissue combined cofractionation experiments in synaptosomes with curation of brain-derived pulldown data and AlphaFold2 modeling to validate tissue-specific associations [45].
Table 3: Essential Research Reagents and Databases for TS-PPI Network Research
| Resource | Type | Function | Application in TS-PPI Studies |
|---|---|---|---|
| BioGRID | Interaction Database | Repository of protein and genetic interactions | Source of experimentally validated PPIs for network construction [22] |
| CORUM | Protein Complex Reference | Curated database of mammalian protein complexes | Ground truth for validating association prediction methods [45] |
| Human Protein Atlas | Expression Database | Tissue and cell type expression profiling | Determining tissue association of proteins [42] [45] |
| Cytoscape | Visualization Software | Network visualization and analysis | Visualizing and analyzing constructed TS-PPI networks [42] |
| STRING | Integrated Database | Functional protein association networks | Source of both known and predicted interactions [42] [49] |
| cBioPortal | Cancer Genomics Portal | Clinical and genomic data integration | Correlating network features with clinical outcomes [49] |
| GTEx | Expression Database | Gene expression across normal human tissues | Reference for normal tissue expression patterns [43] |
| Clue.io | Drug Database | Information on drug-target interactions | Identifying potential therapeutic compounds [49] |
Applications in Disease Research and Drug Development
The application of TS-PPI networks has demonstrated significant value across multiple domains of biomedical research:
Disease Gene Prioritization: Tissue-specific networks outperform generic PPI networks in prioritizing candidate disease-causing genes [42] [45]. For example, in brain disorders, constructing networks of schizophrenia-related genes effectively prioritizes candidates in loci linked to the disease [45].
Cancer Subtype Stratification: APPIC enables identification of consensus PPI networks specific to patient cohorts, revealing subtype-specific mechanisms in 10 cancer types [49]. This approach can identify hub proteins with high connectivity that represent potential therapeutic targets [49].
Toxicology and Substance Use Disorders: Construction of a HUD (Heroin Use Disorder) PPI network identified 111 nodes with 553 edges, with JUN and PCK1 emerging as central components potentially involved in addiction mechanisms [47].
Elucidation of Tissue-Specific Disease Mechanisms: Genes causing hereditary diseases tend to have higher transcript levels and more interacting partners in disease-relevant tissue networks compared to unaffected tissues [42]. This pattern helps explain why mutations in widely expressed genes cause pathology only in specific tissues.
The construction and analysis of tissue and tumor-specific PPI networks represents a critical advancement beyond generic interactome mapping. Tools like SPECTRA, TissueNet v.2, and APPIC provide researchers with sophisticated platforms to explore the context-dependent nature of protein interactions. The integration of high-quality PPI data from sources like BioGRID and IntAct with extensive expression datasets from TCGA, HPA, and GTEx enables creation of biologically realistic network models.
As the field progresses, several emerging trends are shaping future development: the shift from mRNA-based to protein co-abundance association metrics; the integration of single-cell resolution data; the incorporation of structural predictions from AlphaFold2; and the development of dynamic network models that capture interactions across different cellular states. For researchers in drug development, these advances offer increasingly precise maps of disease mechanisms within their relevant physiological contexts, enabling more targeted therapeutic strategies with potentially reduced off-target effects.
The ongoing expansion of both interaction and expression data, coupled with more sophisticated analytical frameworks, promises to further enhance our understanding of how protein networks orchestrate tissue-specific functions and how their dysregulation drives pathological processes in specific tissue contexts.
Protein-Protein Interaction (PPI) networks provide a physical map of the cellular machinery, where nodes represent proteins and edges represent their functional interactions [51] [52]. The mining of functional modules—groups of proteins that work together to carry out specific biological processes—from these complex networks is a fundamental task in systems biology. These modules often correspond to molecular complexes, pathways, or functional units, and their identification is crucial for understanding cellular organization, disease mechanisms, and identifying potential drug targets [53]. The analysis of PPIs has been transformed by the availability of large-scale interaction databases such as BioGRID, which as of November 2025 contained over 2.25 million non-redundant interactions curated from more than 87,393 publications [22], and STRING, which provides both known and predicted interactions for billions of protein pairs [54].
Algorithms for detecting these functional modules largely fall into two categories: traditional methods that optimize edge density between partitions, and structurally-based approaches like the Structural Clustering Algorithm for Networks (SCAN). SCAN offers a unique advantage by identifying not only clusters but also hubs (highly connected proteins linking modules) and outliers (proteins with weak connections), providing a more nuanced view of network topology [53]. This technical guide explores the principles, implementation, and validation of functional module mining, with a specific focus on the SCAN algorithm and its application within the context of modern PPI research resources.
Algorithmic Foundations: SCAN and Its Mechanics
Core Principles of the SCAN Algorithm
The Structural Clustering Algorithm for Networks (SCAN) is a density-based clustering algorithm that identifies clusters, hubs, and outliers in networks based on the structural similarity of vertices. Unlike modularity-based algorithms or normalized cut methods that partition networks to maximize intra-cluster edges and minimize inter-cluster edges, SCAN defines clusters based on the notion that two nodes belong to the same community if they share a similar neighborhood [53]. This approach is particularly suited to biological networks like PPIs, where proteins sharing many interaction partners are likely to be functionally related.
The algorithm's theoretical foundation rests on the observation that many complex biological networks exhibit significant common principles including small-world properties, power-law degree distributions, and highly modular structures [53]. SCAN efficiently detects these modules by leveraging structural similarities, with an empirical analysis demonstrating linear running time relative to network size, making it one of the fastest approaches available for large-scale networks [53].
Key Definitions and Mathematical Formulation
SCAN operates using several key concepts and parameters that determine cluster formation:
- Structural Similarity: For two nodes, v and w, their structural similarity is defined as the normalized number of common neighbors: σ(v,w) = |N(v) ∩ N(w)| / √(|N(v)| · |N(w)|), where N(v) and N(w) are the sets of neighbors of v and w, respectively. This geometric normalization accounts for differences in node degrees.
- ε (Epsilon): A similarity threshold parameter. Two nodes are considered similar if their structural similarity is at least ε.
- μ (Mu): A minimum cluster size parameter that controls the formation of core nodes.
- Core Node: A node v is a core node if it has at least μ neighbors that are similar to it (i.e., with similarity ≥ ε).
- Directly Reachable: A node w is directly reachable from a core node v if w is similar to v.
- Reachable: A node w is reachable from v if there is a path v₁,...,vₙ of core nodes where v₁ = v and w is directly reachable from vₙ.
- Connected Cluster: A cluster is formed by all nodes reachable from its core nodes.
- Hub: A non-core node that neighbors two or more different clusters.
- Outlier: A non-core node that neighbors only one cluster.
Table 1: SCAN Algorithm Parameters and Their Functions
| Parameter | Type | Function in Algorithm | Biological Interpretation |
|---|---|---|---|
| ε (Epsilon) | Continuous (0-1) | Similarity threshold for considering nodes neighbors | Controls cluster granularity; lower values create larger, more inclusive clusters |
| μ (Mu) | Integer ≥ 2 | Minimum number of similar neighbors for core node formation | Determines how well-connected a protein must be to form a cluster core |
| Structural Similarity | Calculated metric | Measures neighborhood overlap between nodes | Quantifies functional relationship between proteins based on shared interactors |
Implementation Workflow: From PPI Data to Functional Modules
Data Acquisition and Preprocessing
The first critical step in functional module mining involves acquiring high-quality PPI data from curated databases. Key resources include:
- BioGRID: A comprehensive repository containing over 2.25 million non-redundant biological interactions as of 2025, with monthly updates ensuring current data [22].
- STRING: Provides both known and predicted protein associations, with coverage exceeding 59 million proteins and 20 billion interactions [54].
- IntAct: Offers molecular interaction data curated from the scientific literature.
Data preprocessing involves cleaning and standardizing the interaction data: removing redundant interactions, handling self-interactions, and standardizing protein identifiers to ensure consistency. For the SCAN algorithm specifically, the network must be represented as an undirected graph G = (V, E), where V is the set of proteins and E is the set of interactions.
SCAN Algorithm Execution Steps
The SCAN algorithm implementation follows a structured process:
Compute Structural Similarity: For each edge (v, w) in the network, calculate the structural similarity σ(v, w). This creates a weighted graph where edge weights represent similarity scores.
Identify Core Nodes: For each node v, check if it has at least μ neighbors with similarity ≥ ε. If so, mark v as a core node.
Depth-First Cluster Expansion: For each unvisited core node v, start a new cluster and recursively add all nodes reachable from v through a chain of core nodes where each consecutive pair has similarity ≥ ε.
Classify Non-Member Nodes: After cluster formation, classify remaining nodes as hubs or outliers based on their connections to the identified clusters.
The following table summarizes the key computational steps and their outputs:
Table 2: SCAN Algorithm Execution Steps and Outputs
| Step | Input | Process | Output |
|---|---|---|---|
| Similarity Calculation | Graph G = (V, E) | Calculate σ(v,w) for all connected node pairs | Weighted graph with similarity scores |
| Core Identification | Weighted graph, parameters ε, μ | Identify nodes with ≥ μ similar neighbors | Set of core nodes |
| Cluster Expansion | Core nodes, similarity graph | Depth-first search from core nodes via similar edges | Preliminary clusters |
| Role Classification | Preliminary clusters, remaining nodes | Analyze connections of non-member nodes | Final clusters, hubs, outliers |
Parameter Optimization and Tuning
Selecting appropriate values for ε and μ is critical for meaningful biological results. Empirical studies suggest:
- ε (Similarity Threshold): Typically ranges from 0.4 to 0.7. Lower values produce larger, more inclusive clusters; higher values create smaller, more tightly related clusters.
- μ (Minimum Cluster Size): Usually set between 2 and 5 for most PPI networks. This parameter helps filter out small, potentially spurious clusters.
Optimal parameter selection can be guided by validation metrics such as clustering score and functional enrichment p-values, discussed in the following section.
Validation and Biological Interpretation
Gene Ontology Enrichment Analysis
The primary method for validating identified functional modules is through Gene Ontology (GO) enrichment analysis. The Gene Ontology database provides controlled vocabularies describing molecular functions, biological processes, and cellular components of gene products [53]. For each cluster, a statistical p-value is calculated to determine if proteins in the cluster are significantly enriched for specific GO terms compared to what would be expected by random chance.
The p-value calculation uses the hypergeometric distribution: p-value = Σ (M choose i)(N-M choose n-i) / (N choose n), where:
- N = total number of proteins in the PPI database
- M = number of proteins in the database with a particular annotation A
- n = cluster size
- i = number of proteins in the cluster sharing annotation A
A cluster is considered significantly enriched with a particular functional annotation if its p-value is below a cutoff threshold (typically 0.05) after multiple testing correction [53].
Validation Metrics and Performance Assessment
To quantify the overall quality of the clustering results, studies often employ a clustering score metric that accounts for both significant and insignificant clusters:
Clustering Score = 1 - [Σ min(pᵢ) + (nᵢ * cutoff)] / [(nₛ + nᵢ) * cutoff]
Where:
- nₛ = number of significant clusters
- nᵢ = number of insignificant clusters
- min(pᵢ) = smallest p-value of a significant cluster
- cutoff = significance threshold (typically 0.05)
In validation studies on yeast PPI networks, SCAN achieved higher clustering scores compared to alternative methods like the CNM (Clauset-Newman-Moore) modularity-based algorithm, particularly for biological process categories [53].
Table 3: Example SCAN Performance on Yeast PPI Network (Saccharomyces cerevisiae)
| Cluster ID | P-Value | GO Term | Term Frequency in Network | Term Frequency in Cluster | Cluster Size |
|---|---|---|---|---|---|
| 1 | 4.45E-98 | nuclear mRNA splicing, via spliceosome | 66 | 58 | 88 |
| 89 | 1.01E-65 | translation | 252 | 58 | 64 |
| 5 | 1.16E-52 | ubiquitin-dependent protein catabolic process | 60 | 34 | 56 |
| 2 | 9.04E-40 | transcription from RNA polymerase II promoter | 50 | 41 | 288 |
| 15 | 8.58E-38 | anaphase-promoting complex-dependent proteasomal ubiquitin-dependent protein catabolic process | 13 | 13 | 13 |
Advanced Validation: Predicting Pathway Membership and PPIs
Beyond GO term enrichment, identified modules can be validated through their ability to predict members of known pathways and protein-protein interactions. Recent approaches have integrated hierarchical constraints from phenotype ontologies to improve module detection. The CMNMF (Consistent Multi-view Nonnegative Matrix Factorization) framework, for instance, factorizes genome-phenome association matrices at consecutive levels of hierarchical phenotype ontologies to mine functional gene modules [55]. This method has demonstrated effectiveness in predicting KEGG pathway members and PPIs in both mouse and human datasets, outperforming conventional clustering approaches [55].
Research Reagent Solutions and Computational Tools
Implementing functional module mining requires both data resources and computational tools. The following table outlines essential research reagents and their applications:
Table 4: Essential Research Reagents and Computational Tools for Functional Module Mining
| Resource/Tool | Type | Primary Function | Application in Module Mining |
|---|---|---|---|
| BioGRID | Database | Repository of protein, genetic, and chemical interactions | Source of curated PPI data for network construction |
| STRING | Database | Known and predicted protein-protein associations | Provides additional interaction context and confidence scores |
| Cytoscape | Software | Network visualization and analysis | Visualization of clusters, hubs, and outliers |
| Gene Ontology | Database | Functional annotation of gene products | Validation of cluster biological significance |
| SCAN Algorithm | Algorithm | Structural clustering of networks | Core methodology for identifying modules, hubs, outliers |
| KEGG Pathways | Database | Collection of pathway maps | Validation of predicted functional modules |
Applications in Drug Discovery and Target Identification
The mining of functional modules from PPI networks has significant implications for drug discovery and target identification. Aberrant PPIs underpin a wide range of human diseases, including neurodegenerative disorders, cancer, and various genetic diseases [51]. Identifying disease-relevant modules can:
Pinpoint Key Therapeutic Targets: Hubs that connect multiple functional modules often represent critical control points in cellular networks. Targeting these proteins may allow modulation of entire disease-relevant pathways.
Reveal Disease Mechanisms: Functional modules disrupted in disease states can reveal the molecular basis of pathology. For example, in neurodegenerative diseases like Alzheimer's, modules containing amyloid-β and tau proteins have been identified [51].
Guide Biologics Design: Sequence-based PPI prediction methods informed by functional module analysis are reshaping drug discovery, particularly in the development of therapeutic peptides and antibodies [51]. Successful examples include the design of peptide binders with nanomolar affinity against targets like NCAM1 and AMHR2 using sequence-based methods that outperformed structure-based approaches [51].
The integration of functional module analysis with drug discovery pipelines represents a powerful approach for identifying and prioritizing novel therapeutic targets in the era of precision medicine.
Protein-protein interactions (PPIs) represent the fundamental regulatory framework governing cellular signaling, transduction, and function in all living organisms. Disruptions in homeostatic PPI patterns provide a direct link between cellular stressors and disease phenotypes, making interactome analysis crucial for understanding pathological mechanisms and identifying therapeutic targets. This technical guide examines integrated approaches for investigating PPI networks, focusing on methodologies from mass spectrometry-based proteomics to advanced computational predictions using deep learning architectures. We explore the transformative potential of the newly developed dysfunctional Protein-Protein Interactome (dfPPI) platform for detecting dynamic changes in PPI networks under disease conditions. By framing our analysis within the context of major PPI databases and resources, this review provides researchers with a comprehensive toolkit for advancing drug discovery through PPI network modulation, supported by detailed experimental protocols, quantitative data summaries, and visual workflow representations.
The Biological Significance of PPIs
Protein-protein interactions form the essential backbone of cellular communication systems, enabling the coordination of complex biological processes including signal transduction, cell cycle regulation, transcriptional control, and metabolic pathway coordination [56] [57]. The human interactome is predicted to encompass between 130,000 and 600,000 distinct interactions, creating an intricate network that maintains cellular homeostasis [56]. These interactions include structural proteins within cells, multi-protein complexes involved in core processes such as transcription and translation, cell-cell adhesion and communication mechanisms, protein synthesis and degradation pathways, and sophisticated signaling cascades [56]. The comprehensive study of PPI networks and the global physical organization of cells provides critical insights into basic cellular biochemistry and physiology, forming a necessary foundation for understanding disease mechanisms.
When the homeostatic state of an organism or individual cell is disturbed due to environmental stress or disease pathology, the normal patterns of PPIs are frequently disrupted [56]. While many such disruptions represent secondary effects with minimal functional consequences, certain PPI disturbances play causal roles in disease initiation and progression [56]. Well-established examples include perturbations in the p53 tumor suppressor interactome caused by genetic mutations, disruptions in desmosome-mediated cellular interactions implicated in various diseases, aberrant PPIs leading to protein aggregate accumulation in neurodegenerative disorders, and host-pathogen PPIs critical in infectious diseases [56]. Consequently, monitoring and analyzing PPIs across different biological models provides significant opportunities for identifying both diagnostic biomarkers and therapeutic targets with broad clinical applicability.
Research into protein-protein interactions relies on numerous expertly curated databases that aggregate interaction data from high-throughput experiments and scientific literature. These resources provide the foundational data for network analysis and disease gene identification. The table below summarizes key databases essential for PPI research.
Table 1: Essential PPI Databases and Resources
| Database | Primary Focus | Data Content | URL |
|---|---|---|---|
| IntAct | Molecular interaction data | Protein-protein interactions curated from literature | https://www.ebi.ac.uk/intact/ |
| BioGRID | Genetic and protein interactions | Protein-protein and genetic interactions from multiple species | https://thebiogrid.org/ |
| STRING | Known and predicted PPIs | Functional protein associations, both direct and indirect | https://string-db.org/ |
| MINT | Experimentally verified PPIs | Protein interactions focused on high-throughput experiments | https://mint.bio.uniroma2.it/ |
| HPRD | Human protein reference | Interaction, enzymatic, and cellular localization data | http://www.hprd.org/ |
| DIP | Experimentally determined PPIs | Catalog of verified protein interactions | https://dip.doe-mbi.ucla.edu/ |
| CORUM | Mammalian protein complexes | Experimentally verified protein complexes | http://mips.helmholtz-muenchen.de/corum/ |
These databases employ different curation methodologies and focus areas, making them complementary resources for researchers. Integration of data from multiple sources often enhances the coverage and reliability of PPI networks for disease analysis [28]. The growing volume of interaction data in these repositories has enabled the development of sophisticated computational approaches for predicting novel interactions and identifying disease-relevant network modules.
Methodological Approaches for PPI Investigation
Experimental Techniques for PPI Mapping
Mass spectrometry-based approaches have revolutionized large-scale mapping of physical interactions to probe disease mechanisms [56]. These methodologies allow for systematic characterization of PPI networks and protein complexes, providing insights into the molecular basis of common diseases including cancer, cardiomyopathies, diabetes, microbial infections, and genetic and neurodegenerative disorders [56]. Several well-established experimental approaches form the cornerstone of PPI research.
Affinity Purification Mass Spectrometry (AP-MS) has emerged as a predominant method for isolating and identifying protein complexes [56]. This approach typically utilizes a tagged 'bait' protein or co-immunoprecipitation with a specific antibody, followed by proteolytic cleavage (usually with trypsin) and MS-based sequencing of resulting peptides to deduce protein identities [56]. When experimental parameters are optimized, AP-MS can reliably detect interactions even for low-abundance proteins, though scaling to hundreds of targets remains challenging [56]. Tandem affinity purification (TAP) and sequential peptide affinity (SPA) tagging technologies have significantly streamlined AP-MS identification and characterization of PPIs and heterogeneous protein complexes [56].
Biochemical Co-fractionation represents an alternative approach involving chromatographic separation of endogenous protein complexes followed by native gel MS identification [56]. This method has proven viable for global profiling of native PPI interaction networks in cell lines, though with generally reduced sensitivity compared to AP-MS approaches [56]. The methodology is particularly valuable for studying membrane-associated complexes and proteins difficult to tag without disrupting function.
Yeast Two-Hybrid (Y2H) Systems continue to provide valuable data, especially for mapping transient interactions that might be missed by MS-based methods [56]. These systems have been used with great success in mapping individual PPIs, including both stable and transient interactions, and complement MS-based methodologies that excel at detecting stably co-purifying multi-component complexes [56].
Table 2: Comparative Analysis of Primary PPI Investigation Methods
| Method | Principle | Advantages | Limitations | Common Applications |
|---|---|---|---|---|
| AP-MS | Affinity purification of tagged bait protein followed by MS identification | High specificity; works for low-abundance proteins; identifies direct and indirect interactions | False positives from sticky proteins; challenging to scale; requires tagging | Systematic mapping of protein complexes; pathway identification |
| Y2H | Reconstruction of transcription factor through protein interaction in yeast | Detects transient interactions; high throughput; measures binary interactions | False positives from auto-activators; limited to non-cytotoxic proteins; nuclear context only | Binary interaction mapping; interaction domain mapping |
| Co-fractionation + MS | Native separation of protein complexes by chromatography | Studies native complexes; no genetic manipulation needed; captures physiological states | Lower sensitivity; complex data analysis; limited dynamic range | Native complex organization; organellar proteomics |
| Cross-linking + MS | Chemical cross-linking of interacting proteins followed by MS | Identifies interaction interfaces; captures transient interactions; provides spatial constraints | Technical complexity; low efficiency; specialized expertise required | Interaction interface mapping; structural modeling |
Experimental Workflow Visualization
The following diagram illustrates a standard integrated workflow for experimental PPI analysis using mass spectrometry-based approaches:
The Scientist's Toolkit: Essential Research Reagents
Successful PPI investigation requires specialized reagents and materials designed for capturing, identifying, and validating protein interactions. The following table details essential research reagents and their applications in PPI studies.
Table 3: Essential Research Reagents for PPI Investigation
| Reagent/Material | Function | Application Examples |
|---|---|---|
| Affinity Tags (TAP, FLAG, HA) | Enable specific purification of bait protein and associated complexes | TAP tagging for sequential purification; FLAG tagging for immunopurification |
| Cross-linkers (Formaldehyde, DSS) | Stabilize transient interactions before purification | Formaldehyde for in vivo cross-linking; DSS for soluble protein complexes |
| Protease Inhibitors | Prevent protein degradation during purification | Complete Mini EDTA-free tablets for maintaining complex integrity |
| Lysis Buffers | Extract proteins while preserving native interactions | RIPA buffer for stringent conditions; NP-40 for mild extraction |
| Antibodies for Co-IP | Specifically immunoprecipitate target proteins | Anti-FLAG M2 agarose for tagged proteins; protein A/G beads |
| Trypsin/Lys-C | Digest proteins into peptides for MS analysis | Sequencing-grade modified trypsin for efficient protein digestion |
| Stable Isotope Labels (SILAC) | Enable quantitative comparison of protein abundance | SILAC kits for quantitative proteomics between conditions |
| Protein Interaction Arrays | High-throughput screening of potential interactions | Human proteome microarrays for interaction partner screening |
Computational and AI-Driven Approaches
Deep Learning Architectures for PPI Prediction
The application of deep learning in computational biology has transformed PPI prediction through its powerful capabilities for high-dimensional data processing and automatic feature extraction [28]. Unlike conventional machine learning algorithms that rely on manually engineered features, deep learning autonomously extracts semantic sequence context information from sequence and residue data, making it particularly well-suited for processing large-scale PPI datasets [28]. Several core architectures have emerged as particularly effective for PPI analysis.
Graph Neural Networks (GNNs) based on graph structures and message passing adeptly capture local patterns and global relationships in protein structures [28]. By aggregating information from neighboring nodes, GNNs generate node representations that reveal complex interactions and spatial dependencies in proteins [28]. Key variants include Graph Convolutional Networks (GCNs), which employ convolutional operations to aggregate information from neighboring nodes; Graph Attention Networks (GAT), which introduce attention mechanisms to adaptively weight neighboring nodes based on relevance; GraphSAGE, designed for large-scale graph processing through neighbor sampling and feature aggregation; and Graph Autoencoders (GAE), which utilize encoder-decoder frameworks to generate compact node embeddings [28].
Convolutional Neural Networks (CNNs) effectively capture local sequence patterns and structural motifs that influence interaction potentials. These architectures process protein sequences or structural features through multiple convolutional layers to identify characteristic patterns associated with binding interfaces. CNNs have demonstrated particular utility when combined with evolutionary information from multiple sequence alignments.
Transformers and Attention Mechanisms have recently been adapted for PPI prediction, leveraging self-attention to capture long-range dependencies in protein sequences and identify potentially interacting residues distant in sequence but proximal in three-dimensional space [28]. The attention mechanisms provide interpretable insights into which residues contribute most significantly to interaction predictions.
Computational Workflow for PPI Prediction
The following diagram illustrates a comprehensive computational workflow for deep learning-based PPI prediction:
Data Integration Frameworks
Multiple data integration represents a powerful methodology for identifying disease genes by collecting evidence from diverse data sources [58]. Markov Random Field (MRF) theory combined with Bayesian analysis provides a flexible framework for incorporating different kinds of biological data, including known gene-disease associations, protein complexes, PPIs, pathways, and gene expression profiles [58]. This approach considers comprehensive characteristics of heterogeneous datasets to capture the complex relationship between genotypes and phenotypes, addressing limitations of methods that only consider direct edges between candidate genes and known disease genes [58].
The MRF-based integration method formulates the disease gene identification problem as a Bayesian labeling problem where the collection of human genes constitutes the site set and disease association status (1 or 0) represents the label set [58]. According to the Hammersley-Clifford theorem, the random field follows a Gibbs distribution, enabling the calculation of posterior probabilities for disease association based on integrated evidence from multiple data sources [58]. This approach has demonstrated strong performance, achieving an AUC score of 0.743 when integrating multiple biological data types in leave-one-out validation experiments [58].
Applications in Disease Mechanism Elucidation
Cancer Research Applications
PPI network analysis has provided transformative insights into cancer mechanisms, particularly through the study of tumor suppressor networks, oncogenic signaling pathways, and therapy resistance mechanisms. The dysfunctional PPI networks in cancer cells create specific dependencies that can be exploited therapeutically [59]. The dfPPI platform has identified dysfunctions integral to maintaining malignant phenotypes and discovered strategies to enhance the efficacy of current therapies [59]. Specific applications include:
Tumor Suppressor Network Analysis: Studies of the p53 interactome have revealed how mutations disrupt normal PPI patterns, leading to uncontrolled cell proliferation and genomic instability [56]. AP-MS approaches have identified SCRIB and ER alpha-interacting proteins in breast cancer, EGFR-associated proteins in lung cancer, and HIF2-interacting proteins in melanoma, providing new insights into tumor-specific vulnerabilities [56].
Oncogenic Signaling Pathways: Mapping of protein complexes in signaling pathways such as PI3K/AKT, RAS/RAF, and WNT/β-catenin has revealed how rewired PPI networks drive oncogenic transformation. Quantitative MS-based approaches have enabled monitoring of interaction dynamics in response to pathway activation or inhibition, providing insights for combination therapies.
Therapy Resistance Mechanisms: Analysis of altered PPI networks in treatment-resistant cancers has identified compensatory interactions that maintain survival signaling despite targeted therapy. These insights have led to strategies for preventing or overcoming resistance through multi-target approaches.
Neurodegenerative Disorders
In neurodegenerative diseases such as Huntington's, Alzheimer's, and prion disorders, PPI network analysis has revealed critical dysfunctions in cellular processes and stressor-specific vulnerabilities [56] [59]. AP-MS identification of huntingtin-interacting proteins has uncovered novel components of pathogenic mechanisms in Huntington's disease [56]. Similarly, study of prion protein interactors has provided insights into the propagation of misfolded proteins in bovine spongiform encephalopathy and related human disorders [56]. Key findings include:
Protein Aggregation Pathways: PPI analysis has identified factors that influence the aggregation propensity of proteins like tau, α-synuclein, and huntingtin, revealing potential intervention points for preventing toxic aggregate formation.
Cellular Quality Control Systems: Mapping interactions within proteostasis networks, including chaperones, ubiquitin ligases, and autophagy components, has revealed how these systems become overwhelmed or dysfunctional in neurodegeneration.
Neuronal Survival Pathways: Identification of protein complexes critical for neuronal survival has provided targets for neuroprotective strategies, with PPI stabilizers offering promising approaches for enhancing the resilience of vulnerable neuronal populations.
Infectious Disease Mechanisms
Microbial pathogens extensively manipulate host PPI networks to establish infection and evade immune responses [56]. The study of host-pathogen PPIs has become central to understanding infectious disease mechanisms and developing novel antimicrobial strategies [56]. AP-MS approaches have systematically identified virus-host PPIs for HIV, measles, hepatitis C, and Japanese encephalitis viruses, revealing how viral proteins rewire cellular networks to support replication [56]. Bacterial pathogens including Borrelia burgdorferi (Lyme disease), Chlamydia trachomatis, Neisseria meningitides, and Staphylococcus aureus have also been studied through PPI analysis of outer membrane protein complexes [56]. Applications include:
Viral Hijacking Mechanisms: Comprehensive mapping of virus-host interactomes has revealed how viral proteins target key cellular complexes to redirect resources toward viral replication and suppress antiviral defense mechanisms.
Bacterial Effector Systems: Analysis of bacterial secretion systems and their protein targets in host cells has illuminated pathogenesis mechanisms and suggested strategies for disrupting critical interactions.
Antimicrobial Discovery: Identification of essential pathogen-specific PPIs has provided new targets for antimicrobial development, with particular promise for agents that disrupt virulence-associated interactions rather than essential bacterial processes.
Drug Discovery Applications
PPI Modulator Development
Protein-protein interactions have transitioned from being considered "undruggable" targets to increasingly feasible therapeutic intervention points, with several FDA-approved drugs now targeting specific PPIs [57]. Approved PPI modulators include maraviroc, tocilizumab, siltuximab, venetoclax, sarilumab, satralizumab, sotorasib, and adagrasib for various diseases including cancer, inflammatory disorders, and viral infections [57]. The development of these agents has demonstrated that despite the challenges posed by typically large and flat PPI interfaces, effective modulation is achievable through multiple strategies:
Small Molecule Inhibitors: These compounds typically target localized "hot spots" within PPI interfaces—regions where a small number of residues contribute disproportionately to binding energy [57]. Hot spots are defined as residues whose substitution results in a substantial decrease in binding free energy (ΔΔG ≥ 2 kcal/mol) and often feature clustered aromatic residues particularly amenable to small molecule binding [57]. Fragment-based drug discovery has proven especially valuable for identifying starting points for PPI inhibitor development, as smaller fragments can bind to subpockets within extensive interface regions [57].
PPI Stabilizers: These represent a more challenging class of modulators that enhance existing protein complexes by binding to specific sites on one or both proteins [57]. Unlike inhibitors that disrupt interfaces, stabilizers often act allosterically and require a profound understanding of the intricate forces governing PPI thermodynamics [57]. The inherent weakness of many PPIs presents additional hurdles for stabilizer development, necessitating innovative approaches for identifying molecules that significantly enhance interaction stability [57].
Peptidomimetics and Macrocyclics: These compounds recapitulate key secondary structure elements of protein interaction domains, particularly α-helices, which represent frequently occurring recognition motifs in PPIs [57]. Computational modeling coupled with phage display technology has enabled rational design of peptidomimetics that maintain bioactive conformations while improving pharmacological properties.
Network Pharmacology and Target Identification
The topological analysis of drug targets within PPI networks provides valuable insights for therapeutic development [60]. Contrary to initial hypotheses, drug targets are neither dominant hub proteins nor critical bridge proteins in PPI networks, but rather occupy distinct topological positions that differ significantly from non-target proteins [60]. Drug targets primarily belong to three typical communities based on their modularity, and these topological features can help predict potential targets or exclude non-targets for efficient drug development [60]. Key principles include:
Network-Based Target Prioritization: Proteins with specific topological characteristics—including particular centrality measures and community affiliations—show enriched likelihood of being druggable targets. These features can be leveraged to prioritize candidates from genome-scale target identification efforts.
Polypharmacology Assessment: Analysis of a drug target's network neighborhood helps predict potential off-target effects and understand multi-target activities that may contribute to efficacy or toxicity.
Therapeutic Window Estimation: The network position of targets relative to essential cellular processes provides insights into potential therapeutic windows, with targets in disease-specific modules often offering better safety profiles.
Therapeutic Development Workflow
The following diagram illustrates the integrated workflow for PPI-targeted therapeutic development:
Emerging Technologies and Future Perspectives
The dfPPI Platform for Systems-Level Investigation
The newly developed dysfunctional Protein-Protein Interactome (dfPPI) platform, formerly known as epichaperomics, represents a significant advancement for detecting dynamic changes at the systems level in PPI networks under stressor-induced cellular perturbations within disease states [59]. This chemoproteomic method provides a direct link between PPI dysfunctions and disease phenotypes, offering a powerful approach for identifying critical vulnerabilities in pathological conditions [59]. The dfPPI platform has particular promise in cancer research, where it identifies dysfunctions integral to maintaining malignant phenotypes and discovers strategies to enhance current therapy efficacy [59]. In neurodegenerative disorders, dfPPI uncovers critical dysfunctions in cellular processes and stressor-specific vulnerabilities [59]. Key advantages include:
Systems-Level Analysis: Unlike approaches focusing on individual interactions, dfPPI captures network-wide perturbations, providing a more comprehensive view of pathological rewiring.
Dynamic Monitoring: The platform can track temporal changes in PPI networks in response to therapeutic interventions, enabling assessment of target engagement and mechanism of action.
Integration Capability: dfPPI data can be combined with other omics datasets to build multi-dimensional models of disease mechanisms and therapeutic responses.
Artificial Intelligence and Machine Learning Advances
Recent advances in deep learning are driving transformative changes in PPI research, with sophisticated architectures enabling increasingly accurate prediction of interactions and characterization of binding mechanisms [28]. The period from 2021 to 2025 has witnessed particularly rapid progress, including attention-driven Transformers, multi-task frameworks, multimodal integration of sequence and structural data, transfer learning via BERT and ESM, and autoencoders for interaction characterization [28]. These approaches have enhanced capabilities for dealing with data imbalances, variations, and high-dimensional feature sparsity that have traditionally challenged computational methods [28]. Promising directions include:
Structure Prediction Integration: The combination of AlphaFold2 and related tools with PPI prediction algorithms enables more accurate modeling of interaction interfaces and the effects of genetic variations.
Multimodal Data Fusion: Integrating diverse data types—including sequence, structure, expression, and functional annotations—through specialized architectures improves prediction accuracy and biological relevance.
Transfer Learning Approaches: Leveraging models pre-trained on large biological datasets enables effective PPI prediction even with limited task-specific training data, particularly valuable for understudied proteins.
Challenges and Future Directions
Despite significant progress, substantial challenges remain in the comprehensive mapping and therapeutic targeting of PPIs. The dynamic nature of interactions, contextual specificity across tissues and conditions, and technical limitations in detecting transient interactions continue to constrain our understanding of interactome networks [57]. Future directions likely to shape the field include:
Time-Resolved Interactomics: Developing methods to capture the temporal dynamics of PPI networks will provide insights into how interactions change during cellular processes, disease progression, and therapeutic interventions.
Single-Cell PPI Analysis: Applying PPI mapping technologies at single-cell resolution will reveal cell-to-cell heterogeneity in interaction networks and identify rare cell population-specific vulnerabilities.
Integration with Clinical Data: Correlating PPI network perturbations with clinical outcomes and treatment responses will strengthen the translational impact of interactome studies and validate therapeutic targets.
PPI Biomarker Development: Identifying disease-specific patterns of PPIs that can serve as biomarkers for improved diagnostics, prognostics, and treatment response monitoring represents a promising application with direct clinical relevance [56].
In conclusion, the integration of experimental and computational approaches for PPI network analysis provides powerful capabilities for elucidating disease mechanisms and identifying therapeutic targets. As technologies continue to advance, particularly in artificial intelligence and chemoproteomics, the systematic investigation of PPIs promises to yield increasingly impactful insights for human health and disease treatment.
Overcoming Common PPI Data Challenges: Noise, Bias, and Validation Techniques
The integration of Protein-Protein Interaction (PPI) data from multiple public databases, such as IntAct and BioGRID, is fundamental to systems biology research, enabling the construction of comprehensive interaction networks. However, this integration is severely hampered by a foundational challenge: the use of inconsistent protein identifiers across different resources. This heterogeneity in data representation creates significant bottlenecks, limiting the reproducibility, scalability, and accuracy of bioinformatics analyses. The problem extends beyond mere inconvenience; it impacts downstream applications in drug discovery and functional genomics, where incomplete or inaccurate networks can lead to flawed biological interpretations.
The core of the issue lies in the independent curation practices and primary data sources of each database. A systematic comparison of 16 human PPI databases revealed that while combined results from STRING and UniHI could cover around 84% of experimentally verified PPIs, no single database provides complete coverage [16]. This finding underscores the necessity of data integration but also highlights the associated challenge of reconciling different identifier schemes, such as those from UniProt, Ensembl, and RefSeq, across these resources. Furthermore, the static nature of PPINs often fails to capture context-specific interactions, a complexity that is lost when identifiers are mapped without considering the biological context of the interaction [61] [62]. This whitepaper provides a technical guide for researchers and drug development professionals to overcome these challenges, outlining robust methodologies for resolving identifier inconsistencies within the context of modern PPI research.
Quantitative Landscape of PPI Database Heterogeneity
Understanding the scope of data heterogeneity requires a quantitative assessment of the coverage and overlap among major PPI databases. A detailed analysis of 16 databases illustrates the fragmented nature of the current PPI data landscape.
Table 1: Experimentally Verified PPI Coverage Across Major Databases
| Database | Description | Human Proteins Covered | Notable Features and Coverage |
|---|---|---|---|
| STRING | Manually curated and predicted interactions | 21,509 (KEGG) | Combined with UniHI, covers ~84% of experimentally verified PPIs [16] |
| UniHI | Unified Human Interactome | N/A | Combined with STRING, covers ~84% of experimentally verified PPIs [16] |
| Reactome | Manually curated biological pathways | 11,442 | Provides detailed information on human biological pathways and processes [63] |
| KEGG Pathway | Maps molecular interaction networks | 21,509 | Covers metabolism, genetic information processing, and human diseases [63] |
| BioGRID | Physical and genetic interactions | N/A | A well-established repository for direct protein interactions [62] |
| IntAct | Molecular interaction database | N/A | Provides a public repository for molecular interaction data [62] |
| hPRINT | Protein-protein interaction network | N/A | Combined with STRING and IID, retrieves ~94% of total PPIs [16] |
| CORUM | Manually curated protein complexes | N/A | Often used as a gold standard, but contains context-specific interactions [61] |
The coverage of specific gene or protein types can also be skewed across databases. For instance, analysis has shown that the coverage of certain databases is uneven for some gene types, meaning that a query for a less-studied protein might yield dramatically different results depending on the database used [16]. This variability is not merely a matter of missing data but is also compounded by the use of different primary identifiers. For example, some databases may use UniProt IDs as their primary key, while others might rely on Ensembl gene IDs or official gene symbols, making automated merging of data a non-trivial task.
Core Strategies for Resolving Identifier Inconsistencies
Resolving protein identifier inconsistencies involves a multi-layered approach, from basic mapping to advanced context-aware integration. The following protocols provide a methodological framework for this process.
Foundational Protocol: Unified Identifier Mapping
The first step in any data integration pipeline is to establish a common identifier system for all proteins. The following workflow is recommended:
- Select a Primary Reference Database: Choose a central database such as UniProt as your standard identifier system. UniProt is widely adopted and provides extensive cross-referencing to other databases.
- Compile Raw PPI Data: Download PPI data from your sources of interest (e.g., BioGRID, IntAct, STRING) using their native identifiers.
- Execute Batch Mapping: Use a programmatic interface to a unified mapping service. The UniProt ID Mapping API is a prime tool for this task. It can take a list of identifiers from various sources (e.g., Ensembl, RefSeq, gene symbols) and return the corresponding UniProt IDs.
- Validate and Handle Ambiguities: Not all identifiers will map perfectly. Manual curation is required for:
- Obsolete Identifiers: Identify proteins that have been removed or merged in the current reference database.
- One-to-Many Mappings: Resolve cases where a single gene symbol maps to multiple UniProt entries (e.g., due to protein isoforms or paralogs). This often requires additional contextual information, such as tissue specificity or sequence data.
- Generate a Unified Interaction Table: Create a final PPI table where all interacting partners are identified with your primary UniProt IDs, replacing all original source-specific identifiers.
Advanced Protocol: Context-Aware Integration Using Heterogeneous Networks
For more sophisticated analyses, such as predicting novel pathways or dynamic properties, a simple ID mapping is insufficient. A heterogeneous network approach can preserve the richness of the original data while enabling integration [63].
- Network Construction: Model the proteomic data as a network where nodes represent proteins. Instead of forcing a single identifier, allow nodes to be annotated with multiple attributes and identifiers from various sources.
- Data Representation (Embedding): Use graph representation learning techniques to transform the high-dimensional network data into a lower-dimensional vector space. Methods like Graph Convolutional Networks (GCNs) or Graph Attention Networks (GATs) can learn embeddings that aggregate features from a node's neighbors [63].
- Techniques: Matrix factorization or random walk methods (e.g., node2vec) can be used to generate these embeddings, which typically are vectors of length ~1000 for each protein node [63].
- Purpose: These embeddings capture the structural and relational characteristics of each protein within the integrated network, effectively creating a context-rich, unified representation that transcends simple identifier mapping.
- Pathway Prediction and Validation: The embedded network can then be used for downstream machine learning tasks. For example, a Deep Graph Network (DGN) can be trained on this unified model to predict sensitivity relationships or novel pathways directly from the PPIN structure, bypassing the need for kinetic parameters that are often unavailable [62].
Table 2: Key Research Reagent Solutions for PPI Data Integration
| Reagent / Tool | Type | Primary Function in Integration | Source/Access |
|---|---|---|---|
| UniProt ID Mapping API | Web Service/ Tool | Batch conversion of protein identifiers from various namespaces to UniProt IDs | UniProt Website |
| BioGRID | PPI Database | Provides physical and genetic interaction data with multiple identifier types | BioGRID Website |
| IntAct | PPI Database | Supplies molecular interaction data; a source for multiple identifier schemes | IntAct Website |
| STRING | PPI Database | Offers integrated interaction data (curated and predicted) for network construction | STRING Website |
| Graph Neural Network (GNN) Libraries (e.g., PyTorch Geometric) | Software Library | Implements GCNs and GATs for creating node embeddings from heterogeneous networks | Open Source (Python) |
| CORUM Subset (Context-Specific) | Gold Standard Dataset | Validates integrated networks and ML predictions under specific experimental conditions | CORUM Website |
Workflow Visualization: From Raw Data to Integrated Network
The following diagram illustrates the end-to-end process of resolving identifier heterogeneity to build a context-aware integrated PPI network.
Experimental Validation and Case Study
Validating the success of an integration pipeline is critical. A recommended method is to use a subset of the CORUM database as a gold standard. Research has identified a specific subset of CORUM complexes that show consistent evidence of interaction in co-fractionation experiments [61]. Using this subset for validation, rather than the entire CORUM database, dramatically improves the accuracy of interactome mapping, as judged by the number of predicted interactions at a given error rate.
Case Study: Validating an Integrated Network for Pathway Analysis
- Objective: Predict novel proteins involved in a specific biochemical pathway.
- Method:
- Integrate PPI data from BioGRID, IntAct, and STRING using the heterogeneous network protocol.
- Train a Graph Neural Network (GNN) model. The model uses the network structure and node embeddings to learn the patterns of known pathway members.
- The GNN then predicts new candidate proteins for the pathway.
- Validation:
- The predictions are tested against the context-specific CORUM gold standard.
- The number of validated interactions at a fixed false discovery rate is compared against a baseline model that used only a single database or simple identifier mapping. The advanced integration method consistently identifies more true positives, demonstrating the value of a robust data harmonization process [61].
The challenge of inconsistent protein identifiers is a significant but surmountable obstacle in PPI research. A strategic approach that combines foundational tools like the UniProt mapping service for basic integration with advanced graph-based machine learning methods for context-aware analysis is essential for building biologically meaningful networks. As the field moves towards more dynamic and condition-specific models of the interactome, the ability to seamlessly integrate diverse data sources will become even more critical. The methodologies outlined in this whitepaper provide a roadmap for researchers to achieve this, thereby enhancing the reliability and power of their research in drug development and systems biology.
Mitigating False Positives and False Negatives in High-Throughput Datasets
In the realm of high-throughput screening (HTS), particularly in protein-protein interaction (PPI) studies and early drug discovery, the reliability of results is critically threatened by two types of errors: false positives and false negatives. A false positive occurs when a test incorrectly indicates the presence of a specific property or interaction when it does not actually exist. Conversely, a false negative occurs when a test fails to detect a genuine interaction or effect that is present. The impact of these errors is particularly pronounced in imbalanced datasets where one class significantly outweighs the other, such as in binary classification scenarios where models tend to favor the majority class, resulting in inadequate predictions for the minority class [64].
The consequences of these errors extend beyond mere statistical inaccuracies. In practical applications such as drug discovery, false positives can lead researchers down unproductive pathways, wasting substantial time and resources. For example, in high-throughput screening systems, over 95% of positive results can be attributed to false positives or unexpected outcomes derived from shared physicochemical properties or interfering factors [65]. Meanwhile, false negatives can cause researchers to overlook potentially valuable interactions or therapeutic compounds, thereby missing critical opportunities for scientific advancement. This is especially critical in fields like medical diagnosis, where a false negative could mean a missed disease diagnosis, or in intrusion detection, where it represents an undetected security breach [64].
Understanding the Core Challenges
False positives in high-throughput screening often arise from various assay interference mechanisms. Common problems include:
- Colloidal aggregation: Where compounds form aggregates that non-specifically inhibit protein activity.
- Spectroscopic interference: Including autofluorescence and compounds that inhibit reporter enzymes like firefly luciferase.
- Chemical reactive interference: Where compounds react covalently with protein targets rather than through specific, reversible interactions.
- Non-specific binding: Compounds binding to targets through non-physiological mechanisms [65].
These interference mechanisms have been recognized as a significant problem in the scientific community. As noted in a commentary by Baell titled 'Chemistry: Chemical con artists foil drug discovery' published in Nature, assay interferent compounds can severely impede the drug development process, leading to substantial waste of research time and resources [65]. The editors-in-chief of American Chemical Society journals further emphasized the harm caused by false-positive compounds in a paper titled 'The Ecstasy and Agony of Assay Interference Compounds,' advising researchers to remain vigilant against potential false positives and confirm the authenticity of positive screening results [65].
False negatives present an equally challenging problem in high-throughput datasets. In machine learning contexts, false negatives often occur in imbalanced classification problems where models excel in predicting the majority class but struggle to identify instances from the minority class [64]. Knowles et al. (2023) draw attention to a tendency in the artificial intelligence domain to underestimate the impact of false negatives, which could have adverse consequences for decision-making, risk assessment, and broader concerns related to the trustworthiness of AI systems [64].
In experimental settings, false negatives can arise from:
- Compounds with poor ionization efficiency in mass spectrometry-based screening methods.
- Insufficient assay sensitivity to detect weak but biologically relevant interactions.
- Technical variability in high-throughput experimental procedures.
- Proteoform-specific interactions that may be missed in bulk screening approaches, particularly relevant in PPI studies where different molecular variants of proteins arising from alternative splicing or genetic variations can significantly influence interaction dynamics and specificity [66].
Computational Mitigation Strategies
Machine Learning Approaches for Imbalanced Datasets
Addressing false negatives in imbalanced datasets requires specialized machine learning approaches. The MinFNR (Minimize False Negative Rate) ensemble algorithm represents a strategic approach to this challenge. This algorithm is designed specifically to minimize False Negative Rates in imbalanced datasets by strategically combining data-level, algorithmic-level, and hybrid-level approaches [64].
Central to the MinFNR algorithm is the use of the Set Covering Problem (SCP), a classic optimization problem that seeks to find the smallest subset of sets that covers all elements in a given universe. For MinFNR, the SCP selects the most relevant classifiers from a pool of candidates, ensuring that all positive instances are correctly identified while minimizing the number of classifiers used [64]. The algorithm works by:
- Training multiple base algorithms on the training dataset using a combination of Data Level, Algorithm Level, and Hybrid approaches.
- Evaluating performance on the test dataset to find performance measures, particularly focusing on Area Under the ROC Curve (ROC-AUC) and FNR.
- Applying the Set Covering Problem to identify the optimal subset of classifiers that collectively minimize the FNR.
- Creating an ensemble from the selected classifiers that provides improved prediction results for highly imbalanced datasets [64].
Through comprehensive evaluation on diverse datasets, MinFNR has consistently outperformed individual algorithms, showing particular potential for applications where the cost of false negatives is substantial, such as fraud detection and medical diagnosis [64].
Specialized Tools for False Positive Identification
For false positive mitigation in drug discovery, specialized computational tools have been developed. ChemFH represents an integrated online platform that facilitates rapid virtual evaluation of potential false positives. This comprehensive tool screens for multiple categories of problematic compounds, including:
- Colloidal aggregators
- Spectroscopic interference compounds
- Firefly luciferase inhibitors
- Chemical reactive compounds
- Promiscuous compounds
- Other assay interferences [65]
The platform leverages a dataset containing 823,391 compounds and employs high-quality prediction models using multi-task directed message-passing network (DMPNN) architectures combined with uncertainty estimation, yielding an average AUC value of 0.91 [65]. Additionally, ChemFH incorporates 1,441 representative alert substructures derived from collected data and ten commonly used frequent hitter screening rules, providing a multi-layered approach to false positive identification.
Table 1: Computational Strategies for Mitigating False Positives and Negatives
| Strategy | Mechanism | Best Suited For | Key Advantages |
|---|---|---|---|
| MinFNR Algorithm | Ensemble method using Set Covering Problem to select classifiers that minimize false negatives | Imbalanced datasets where false negatives have high cost | Specifically targets false negative reduction; combines multiple approaches |
| ChemFH Platform | DMPNN models with substructure alerts to identify frequent hitters | Drug discovery HTS; virtual screening | Covers multiple interference mechanisms; high AUC (0.91) |
| Multi-task DMPNN | Neural network architecture that learns shared representations across related tasks | Large-scale compound screening | Leverages shared information across tasks; improved performance |
| Structure-Based Filters | Pre-defined substructure rules to flag problematic compounds | Initial compound triage | High interpretability; computationally efficient |
Experimental Methodologies for Error Reduction
LC-MS Workflow Eliminating Both False Positives and Negatives
A novel mass spectrometry-based HTS workflow has been developed that uniquely addresses both false positives and false negatives simultaneously. This method, described by researchers in Scientific Reports, employs a reporter displacement approach that eliminates both error types through careful experimental design [67].
The protocol involves the following key steps:
Protein Immobilization: Target proteins are immobilized onto appropriate solid supports. For example, carbonic anhydrase and pepsin can be immobilized onto Aminolink Plus coupling resin, while maltose binding protein can be immobilized on N-hydroxysuccinimide-activated magnetic beads. Proteins are maintained at their optimal pH throughout immobilization [67].
Reporter Molecule Incubation: The immobilized proteins are incubated with a known ionizable weak binder (reporter molecule). For carbonic anhydrase, this could be methoxzolamide; for pepsin, pepstatin A derivatives work effectively.
Library Compound Exposure: The protein-reporter complex is exposed to a batch of library compounds (typically 100-400 compounds per batch), while an equimolar amount of the complex without library compounds serves as a control.
LC-MS Analysis: Liquid chromatography-mass spectrometry is used to detect the reporter molecule. If a stronger binder is present in the library, the signal of the reporter molecule increases compared to the control samples, indicating displacement by a higher-affinity ligand.
Hit Identification: Compounds causing significant reporter displacement are identified as true binders, regardless of their own ionization potential [67].
This method's key innovation lies in detecting binding events through the displacement of a well-ionizing reporter molecule, rather than through direct detection of binding compounds. This approach eliminates false negatives caused by poor compound ionization and minimizes false positives by requiring specific displacement rather than mere detection [67].
Research Reagent Solutions for Robust Screening
Table 2: Essential Research Reagents for Mitigating False Results in High-Throughput Screening
| Reagent/Resource | Function in False Result Mitigation | Application Context |
|---|---|---|
| Aminolink Plus Coupling Resin | Immobilizes target proteins for reporter displacement assays | LC-MS HTS workflow; reduces non-specific binding |
| NHS-Activated Magnetic Beads | Alternative protein immobilization platform; enables rapid separation | Screening low-affinity binders; MBP studies |
| Reporter Molecules | Well-ionizing weak binders that signal displacement by stronger binders | Eliminates false negatives from poor ionization |
| Non-ionic Detergents | Disrupt colloidal aggregates that cause false positives | Counterscreening for aggregation-based interference |
| Scavenging Reagents | Quench reactive compounds that cause false positives | Counterscreening for reactivity-based interference |
| AlphaFold2 Structural Predictions | Provides protein structural data for feature extraction in ML models | PPI prediction; identifying binding interfaces |
Data Integration and Validation Frameworks
Within the context of PPI research, resources like BioGRID provide essential ground truth data for developing and validating computational models. BioGRID serves as a comprehensive repository of biologically relevant PPIs, containing curated data from thousands of publications [22]. As of late 2025, BioGRID contains over 2.2 million non-redundant interactions curated from more than 87,000 publications, making it one of the most extensive PPI databases available [22].
The application of BioGRID data in mitigating false positives and negatives includes:
Benchmark Dataset Creation: High-confidence interaction data from BioGRID can be used to create reliable benchmark datasets for training machine learning models to predict PPIs, particularly in rice and other less-studied organisms [66].
Negative Sample Selection: BioGRID's comprehensive coverage helps in selecting true negative samples—protein pairs that are unlikely to interact based on their presence in different subcellular compartments or lack of any documented interaction across extensive curation efforts.
Homology-Based Inference: For organisms with limited direct PPI data, BioGRID enables homology-based inference from well-studied model organisms. Approximately 40% of Arabidopsis PPIs show detectable conservation in rice, providing a valuable resource for expanding interaction networks [66].
Validation of Screening Results: New interactions identified through high-throughput screens can be cross-referenced with BioGRID to assess novelty and potential biological relevance.
Machine Learning Feature Engineering for PPI Prediction
Effective machine learning approaches for PPI prediction employ sophisticated feature engineering strategies to minimize false predictions:
Sequence-Based Features:
- Amino acid composition and dipeptide frequencies
- Position-Specific Scoring Matrix (PSSM) profiles
- Evolutionary conservation scores
- Physicochemical properties of residues
Structure-Based Features (increasingly available through AlphaFold predictions):
- Secondary structure elements
- Solvent accessibility surface areas
- Electrostatic potential distributions
- Binding site similarity metrics [66]
Network-Based Features:
- Gene co-expression patterns (from resources like RiceFREND)
- Functional annotation similarities
- Domain interaction probabilities
- Phylogenetic profile correlations
The integration of these diverse feature types enables more accurate PPI prediction, significantly reducing both false positives and false negatives compared to single-modality approaches.
Mitigating false positives and false negatives in high-throughput datasets requires a multi-faceted approach combining computational strategies, experimental innovations, and robust data resources. The MinFNR algorithm provides a powerful framework for addressing false negatives in imbalanced classification problems, while tools like ChemFH offer comprehensive solutions for identifying false positives in drug screening. The reporter displacement LC-MS method represents a significant experimental advancement that virtually eliminates both error types through clever assay design.
Looking forward, several emerging trends promise to further enhance our ability to mitigate false results in high-throughput datasets:
Integration of Multi-omics Data: Combining proteomic, transcriptomic, and structural data will provide more contextual information for distinguishing true interactions from artifacts.
Advanced Deep Learning Architectures: Transformer-based models and graph neural networks show promise for more accurate PPI prediction through better representation of biological context.
Uncertainty Estimation in Predictive Models: Incorporating uncertainty quantification, as demonstrated in the ChemFH platform, allows researchers to assess confidence in predictions and prioritize experimental validation efforts.
Proteoform-Aware Interaction Mapping: Accounting for different protein variants and modifications will reduce false negatives caused by interaction specificity to particular proteoforms.
As high-throughput technologies continue to evolve and generate increasingly large datasets, the methods described in this technical guide will become ever more essential for extracting meaningful biological insights from the noise of potential artifacts and missed interactions.
Tackling Biases in Literature-Curated Data and Computational Predictions
Protein-protein interaction (PPI) data forms the foundation for understanding cellular machinery, signaling pathways, and identifying novel therapeutic targets. Resources like BioGRID and IntAct represent cornerstone repositories that compile experimentally verified interactions from thousands of scientific publications [22] [24]. As of late 2025, BioGRID alone contains non-redundant interactions from over 87,000 publications, encompassing more than 2.25 million curated interactions [22]. Despite this impressive scale, both literature-curated and computationally predicted PPI data remain susceptible to multiple forms of bias that can significantly impact research outcomes and biological interpretations.
These biases manifest systematically across the data lifecycle. Experimental bias arises from the predominant use of certain laboratory techniques, while curation bias emerges from human decision-making during data extraction from literature. Computational bias affects predicted interactions through algorithmic limitations and training data composition. Understanding and mitigating these biases is particularly crucial for drug development professionals who rely on accurate PPI networks to identify promising therapeutic targets, as biased data can lead to wasted resources and failed clinical trials.
Experimental and Technological Biases
Experimental methods for determining PPIs each carry inherent technical biases that influence which interactions are detectable. Yeast two-hybrid (Y2H) systems primarily identify binary interactions but may miss complexes requiring post-translational modifications or specific cellular conditions [24]. Affinity purification followed by mass spectrometry (AP-MS) detects protein complexes but struggles to distinguish direct from indirect interactions, leading to representation differences based on whether the "matrix" or "spokes" model is applied during data interpretation [24]. High-throughput methods generate scale at the cost of higher false-positive rates compared to low-throughput focused studies, creating volume imbalances in curated databases.
Literature Curation and Annotation Biases
Human curators introduce biases during data extraction from scientific literature. Confirmation bias may lead to preferential extraction of interactions that align with established biological knowledge, while annotation inconsistencies create integration challenges across databases [24] [68]. Studies comparing multiple PPI databases have found that different databases may report significantly different interaction counts from the same original publication [24]. For example, one analysis found that of 14,899 publications shared by at least two databases, 5,782 (39%) were reported with different numbers of interactions across databases [24]. This problem is compounded by identifier mapping issues where proteins may be inconsistently identified across databases, leading to integration challenges.
Computational Prediction Biases
Deep learning approaches for PPI prediction, including Graph Neural Networks (GNNs), Convolutional Neural Networks (CNNs), and Transformers, have revolutionized our ability to predict interactions at scale [28]. However, these methods inherit and potentially amplify biases present in their training data. Models trained on limited organism data fail to generalize well to less-studied species, creating representation bias [28]. The "bias in, bias out" paradigm is particularly problematic, where models trained on historically biased experimental data perpetuate and amplify these biases in their predictions [68]. Data imbalance issues, where certain protein families have disproportionately more known interactions, lead to models with better performance on well-characterized proteins at the expense of less-studied ones [28].
Table 1: Major Sources of Bias in PPI Data
| Bias Category | Specific Bias Types | Impact on PPI Data |
|---|---|---|
| Experimental | Technique-specific limitations (Y2H, AP-MS) | Preferential detection of certain interaction types |
| High-throughput vs. low-throughput | Volume and quality imbalances | |
| Curation | Confirmation bias | Under-representation of novel interactions |
| Annotation inconsistency | Integration challenges across databases | |
| Computational | Training data representation | Poor performance on less-studied proteins |
| Algorithmic limitations | False positives/negatives with specific patterns |
Methodologies for Bias Assessment and Mitigation
Experimental Design for Bias Evaluation
Systematically evaluating bias requires carefully designed assessment protocols. The following methodology adapts principles from systematic database comparisons to quantify coverage gaps and representation biases:
Protocol 1: Database Coverage Assessment
- Query Set Selection: Curate a balanced set of query genes including both well-studied and under-studied proteins, with representation across different functional classes and expression patterns [16].
- Multi-Database Query: Execute identical queries across multiple PPI databases (BioGRID, STRING, IntAct, etc.) using standardized identifiers and parameters [16].
- Interaction Categorization: Classify returned interactions as experimentally verified (subcategorized by method) or computationally predicted.
- Coverage Calculation: Compute the percentage of unique interactions identified by each database and their overlaps. Studies have found that combined use of STRING and UniHI covers approximately 84% of experimentally verified PPIs, while hPRINT, STRING, and IID together retrieve about 94% of total available interactions [16].
- Gap Analysis: Identify interactions exclusive to each database and analyze their characteristics to detect curation biases.
Protocol 2: Gold-Standard Validation
- Reference Set Compilation: Create a "gold-standard" set of literature-curated, experimentally proven PPIs with particular focus on interactions validated by multiple methods [16].
- Recall Assessment: Measure the percentage of gold-standard interactions recovered by each database or prediction tool. Research indicates that GPS-Prot, STRING, APID, and HIPPIE each cover approximately 70% of curated interactions in benchmark assessments [16].
- Precision Estimation: Evaluate verification rates for database-specific interactions through manual literature checks or experimental validation.
Computational Bias Mitigation Strategies
Advanced computational approaches offer promising pathways for addressing biases in PPI prediction:
Multi-Modal Integration combines evidence from diverse data sources including protein sequences, structural data, gene expression patterns, and functional annotations to reduce over-reliance on any single evidence type [28]. Graph Neural Networks with attention mechanisms (like GAT) can adaptively weight different interaction evidence based on reliability [28]. Transfer Learning approaches using protein language models (ESM, ProtBERT) pre-trained on universal sequence properties help reduce organism-specific biases [28]. Adversarial De-biasing employs discriminator networks to identify and penalize model dependencies on spurious biased correlations in the training data [69].
For database curation, algorithmic auditing frameworks can systematically flag potential biases. The following workflow illustrates an automated bias detection pipeline:
Table 2: Bias Mitigation Strategies for PPI Research
| Mitigation Approach | Implementation Method | Applicable Bias Types |
|---|---|---|
| Multi-Database Integration | Combined use of STRING, UniHI, BioGRID, IID | Coverage bias, Curation bias |
| Multi-Modal Deep Learning | GNNs integrating sequence, structure, expression data | Experimental technique bias |
| Transfer Learning | Pre-training on model organisms, fine-tuning on less-studied species | Organism representation bias |
| Adversarial De-biasing | Fairness-aware adversarial perturbation (FAAP) | Historical bias, Representation bias |
| Data Augmentation | Synthetic data generation for underrepresented classes [69] | Data imbalance, Representation bias |
Experimental Protocols for Bias Validation
Targeted Experimental Verification
Experimental validation remains the ultimate standard for verifying interaction reliability and identifying false positives/negatives resulting from biases. The following protocols provide methodologies for bias-focused verification:
Protocol 3: Coverage Gap Validation
- Objective: Experimentally test interactions consistently missing from major databases but predicted by computational models.
- Method Selection: Choose appropriate experimental methods based on the interaction context. For binary interactions, use Yeast Two-Hybrid with proper positive and negative controls. For complex associations, employ Co-Immunoprecipitation (Co-IP) followed by Western blotting [24] [70].
- Experimental Controls: Include known positive and negative interaction pairs in each experiment to validate methodological effectiveness.
- Documentation: Record all experimental parameters, including expression systems, detection methods, and quantification approaches.
Protocol 4: Orthogonal Method Verification
- Objective: Confirm interactions detected by only one experimental method using complementary techniques.
- Design: Select interactions reported exclusively by either Y2H or AP-MS approaches for verification by orthogonal methods.
- Implementation: Test Y2H-derived interactions using surface plasmon resonance (SPR) to measure binding affinity, and verify AP-MS findings through protein complementation assays (PCA) or FRET-based methods [24].
- Quantification: Employ quantitative measures (e.g., binding constants, interaction scores) to facilitate comparison across methods.
The Researcher's Toolkit for Bias-Aware PPI Research
Table 3: Essential Research Reagents and Resources for Bias Mitigation
| Resource Category | Specific Examples | Application in Bias Mitigation |
|---|---|---|
| Database Platforms | BioGRID, IntAct, STRING, APID, IID | Multi-source data integration, Coverage gap analysis |
| Computational Tools | GNN architectures (GCN, GAT), ESM-2, AlphaFold-Multimer | De-biased prediction, Multi-modal evidence integration |
| Experimental Systems | Yeast Two-Hybrid kits, Co-IP reagents, Proximity ligation assays | Orthogonal validation, Coverage gap verification |
| Reference Sets | Literature-curated gold standards, Negatome (non-interacting pairs) | Method benchmarking, Bias quantification |
| Analysis Frameworks | IMEx standards, PSI-MI data formats, Custom bias audit scripts | Standardized comparison, Systematic bias assessment |
Addressing biases in literature-curated data and computational predictions requires a multi-faceted approach combining computational innovation, rigorous experimental design, and standardized data curation practices. The integration of multi-database queries, systematic bias auditing protocols, and orthogonal experimental validation creates a robust framework for identifying and mitigating biases in PPI research. For drug development professionals, adopting these bias-aware approaches is particularly critical, as biased PPI networks can lead to erroneous target identification and costly late-stage failures. Future directions should include the development of standardized bias reporting standards for PPI databases, increased emphasis on under-studied proteomes, and continued advancement of de-biased computational algorithms. Through collaborative efforts across the research community, we can progressively reduce these biases, leading to more accurate biological models and more successful translation to therapeutic applications.
Protein-protein interaction (PPI) data serves as a critical foundation for understanding cellular mechanisms, disease pathways, and drug target identification. Resources like BioGRID and IntAct provide meticulously curated repositories that support these research endeavors. The reliability of biological discoveries hinges directly on the implementation of rigorous, routine data quality checks throughout the curation and validation lifecycle. For PPI data, quality is multidimensional, encompassing accuracy, completeness, consistency, and experimental validity.
The BioGRID database exemplifies this practice, housing over 1.93 million manually curated interactions from more than 63,000 publications as of 2020 [2]. This vast repository is built upon a structured framework of controlled vocabularies and experimental evidence codes, ensuring that each interaction is traceable to primary experimental data. Manual curation converts unstructured information from text, figures, and tables into standardized, computable records, forming a high-confidence network for the research community [2] [11]. This process transforms raw literature into a refined knowledge resource, enabling complex network analyses and systems biology modeling that drive biomedical discovery.
Core Principles of PPI Data Validation
Validation and verification protocols for PPI data must address distinct types of interactions and their associated challenges. The core principles revolve around experimental traceability, methodological appropriateness, and contextual biological accuracy.
Defining Interaction Types and Evidence
PPIs are fundamentally categorized as either stable or transient, and can be further classified as physical or genetic [71]. Stable interactions, such as those in multi-subunit complexes, are more readily isolated through standard biochemical methods. In contrast, transient interactions, which govern most cellular signaling and regulatory processes, are temporary and often require stabilization techniques like cross-linking for analysis [71]. BioGRID meticulously captures this diversity through structured evidence codes that distinguish between 17 different protein interaction methods (e.g., affinity capture-mass spectrometry, co-crystal structure, two-hybrid) and 11 genetic interaction methods (e.g., synthetic lethality, synthetic rescue) [2].
The Validation Cycle
A robust quality framework implements checks throughout the data lifecycle. The cycle begins with input validation during curation, where experimental details are captured using standardized ontologies. This is followed by methodological verification to ensure the appropriate experimental approach was used to detect the reported interaction type. Contextual validation assesses biological plausibility against existing knowledge, while computational checks identify anomalies through network analysis. Finally, community feedback mechanisms allow for continuous refinement, creating an iterative process that maintains data integrity over time [2] [11].
Experimental Validation Protocols
Routine quality checks require understanding the fundamental experimental methods that generate primary PPI data. Each technique has specific strengths, limitations, and appropriate applications that curators and researchers must recognize when validating interactions.
Table 1: Key Experimental Methods for PPI Validation
| Method | Detection Principle | Suitable for Screening | Affinity/Kinetics Data | Key Applications |
|---|---|---|---|---|
| Pull-Down Assay [72] [71] | Affinity purification with tagged bait protein | Yes | No | Validating direct interaction between known proteins in vitro |
| Yeast Two-Hybrid (Y2H) [72] [73] | Reconstitution of transcription factor via bait-prey interaction | Yes | No | Genome-wide screening for novel nuclear interactions |
| Co-Immunoprecipitation (Co-IP) [72] [71] | Antibody-mediated capture of bait and endogenous prey | No | No | Confirming in vivo interactions in native cellular context |
| FRET [72] | Non-radiative energy transfer between fluorophores | No | Yes (KD) | Real-time dynamics of interactions in live cells (1-10 nm range) |
| ITC [72] | Precise measurement of heat change during binding | No | Yes (KD) | Label-free thermodynamic profiling in solution |
| Surface Plasmon Resonance (SPR) [72] | Detection of mass change on sensor surface | No | Yes (Kon, Koff) | Kinetic characterization of binding events |
Standardized Experimental Workflows
Implementing consistent laboratory protocols is essential for generating reliable, reproducible PPI data. The following workflows represent core methodologies.
Co-Immunoprecipitation (Co-IP) Protocol
Co-IP is a cornerstone method for verifying interactions under native cellular conditions [71]. The protocol involves: 1. Cell Lysis: Preparing lysates under non-denaturing conditions to preserve native protein complexes. 2. Antibody Binding: Incubating the target protein (bait) antibody with the cell lysate. 3. Immobilization: Capturing the antibody-antigen complex using Protein A/G magnetic or agarose beads. 4. Washing: Removing non-specifically bound proteins with gentle buffers. 5. Elution & Analysis: Releasing bound proteins (both bait and prey) for detection by SDS-PAGE and Western blotting [71]. This process confirms that interactions occur in a physiological context, though it cannot distinguish between direct and indirect connections.
Pull-Down Assay Protocol
For in vitro interaction studies, pull-down assays provide a direct approach [72] [71]. The standardized workflow includes: 1. Bait Immobilization: Purifying and binding a tagged bait protein (GST, polyHis, or streptavidin) to appropriate affinity beads. 2. Incubation: Mixing the immobilized bait with a cell lysate or purified prey protein solution. 3. Capture: Allowing potential binding partners to interact with the bait. 4. Washing: Removing unbound proteins to reduce background noise. 5. Elution: Releasing the captured complexes using competitive ligands or specific buffer conditions. 6. Detection: Identifying the precipitated prey proteins through Western blotting or mass spectrometry [71]. This method is particularly valuable for confirming direct binary interactions.
Computational Verification Frameworks
Computational approaches provide essential scalability for quality assessment, complementing experimental validation. These frameworks analyze interaction data from multiple dimensions to assign confidence scores and identify potential anomalies.
Confidence Scoring with Machine Learning
The SPOC (Structure Prediction and Omics-Informed Classifier) framework represents a cutting-edge approach to computational validation [74]. Developed to address the high false-positive rate in proteome-wide AlphaFold-Multimer (AF-M) predictions, SPOC uses machine learning on curated datasets to effectively separate true and false PPI predictions. The classifier integrates structural prediction metrics with orthogonal biological evidence, enabling reliable large-scale interaction screening. This method has been successfully applied to nearly 300 human genome maintenance proteins, generating ∼40,000 high-confidence predictions available through the Predictomes database [74].
Hierarchical Network Analysis
The HI-PPI (Hyperbolic graph convolutional network and Interaction-specific learning for PPI prediction) framework demonstrates how incorporating biological hierarchy improves prediction quality [75]. This deep learning method integrates hyperbolic geometry with graph convolutional networks to capture the natural hierarchical organization of PPI networks, where proteins are organized into functional groups, complexes, and pathways. By representing this hierarchy in hyperbolic space—where the distance from the origin reflects a protein's position in the hierarchy—HI-PPI achieves significant improvements in prediction accuracy, outperforming previous state-of-the-art methods by 2.62%–7.09% in Micro-F1 scores on benchmark datasets [75].
Table 2: Computational Tools for PPI Quality Assessment
| Tool/Resource | Primary Function | Key Features | Data Output |
|---|---|---|---|
| SPOC Classifier [74] | Filters AlphaFold-Multimer predictions | Machine learning based on structure and omics data | Binary classification (true/false) with confidence scores |
| HI-PPI [75] | Predicts and validates PPIs | Hyperbolic geometry captures network hierarchy | Interaction probabilities with hierarchical relationships |
| BioGRID Curation [2] [11] | Manual expert curation | Standardized evidence codes (17 physical, 11 genetic) | Curated interactions with experimental annotation |
Implementing Routine Quality Control Protocols
Establishing systematic quality control (QC) protocols ensures ongoing data integrity in PPI databases and research workflows. These protocols combine automated checks with expert review in a continuous cycle.
Curation and Annotation Standards
BioGRID implements a sophisticated curation pipeline managed through its Interaction Management System (IMS) [11]. This system administers triaged publication lists for curation, ensuring comprehensive coverage. The core QC protocol includes: 1. Literature Triage: Identifying relevant publications through keyword searches and text-mining. 2. Experimental Annotation: Manually curating all protein, genetic, and chemical interactions using controlled vocabularies. 3. Evidence Coding: Tagging each interaction with specific experimental methods (e.g., affinity capture-MS, synthetic lethality). 4. Themed Curation Projects: Conducting focused curation on critical biological areas like the ubiquitin-proteasome system, chromatin modification, and SARS-CoV-2 interactions [2]. This multi-layered approach maintains consistency across thousands of publications and millions of interactions.
Integrated Validation Framework
A comprehensive QC strategy integrates multiple validation approaches, as visualized in the following framework:
This integrated framework ensures that PPI data passes through multiple validation filters before being classified as high-confidence. Automated checks verify basic formatting and completeness, while experimental validation assesses methodological appropriateness. Contextual analysis evaluates biological plausibility within known networks, and computational verification applies statistical and machine learning approaches. Finally, expert review provides the essential human judgment based on domain knowledge.
Essential Research Reagent Solutions
Implementing these validation protocols requires specific research tools and reagents. The following table catalogues essential materials for PPI research, drawn from standardized experimental methods.
Table 3: Essential Research Reagents for PPI Validation
| Reagent/Tool | Primary Function | Application Examples |
|---|---|---|
| Affinity Beads (Protein A/G, Glutathione, Metal Chelate) [71] | Immobilization of bait proteins or antibodies | Co-IP, pull-down assays, complex purification |
| Tag Systems (GST, polyHis, Streptavidin) [71] | Bait protein labeling and purification | Pull-down assays, protein expression and purification |
| Crosslinkers [71] | Stabilization of transient interactions | Capturing weak/transient complexes for analysis |
| Protease Inhibitors [71] | Preservation of protein integrity during lysis | All cell-based preparation methods |
| Plasmid Vectors (GAL4-AD/BD) [72] [73] | Bait and prey expression in two-hybrid systems | Y2H screening, binary interaction testing |
| Fluorescent Proteins (YFP, CFP, mNeonGreen) [73] | Fusion tags for visualization and FRET | Live-cell imaging, interaction dynamics, BiFC |
| CRISPR/Cas9 Systems [2] [11] | Genome editing for validation | Gene knockout, functional validation of interactions |
Maintaining high-quality PPI data requires an integrated, multi-layered approach combining experimental rigor with computational intelligence. As interaction databases continue to grow—with BioGRID now incorporating CRISPR screen data through its ORCS extension—the implementation of systematic validation protocols becomes increasingly critical [2] [11]. The framework presented here, encompassing standardized experimental methods, machine learning classification, hierarchical network analysis, and routine quality control checks, provides a roadmap for ensuring data reliability. For researchers and drug development professionals, adhering to these protocols enhances the translational potential of PPI data, enabling more accurate network modeling, better target identification, and ultimately, more successful therapeutic development.
Leveraging Machine Learning and Transformer Models for Enhanced PPI Extraction from Literature
Protein-Protein Interactions (PPIs) are fundamental regulators of virtually all cellular processes, from signal transduction and immune response to cell cycle progression and transcriptional regulation [28]. The systematic identification of PPIs is therefore crucial for understanding cellular machinery and disease mechanisms, with aberrant PPIs being key contributors to conditions such as neurodegenerative disorders and cancer [51]. Traditional experimental methods for PPI identification, including yeast two-hybrid screening and co-immunoprecipitation, are resource-intensive, time-consuming, and challenging to scale [28]. While public databases such as IntAct, BioGRID, and STRING consolidate PPI information, their coverage is incomplete, and a significant amount of interaction data remains buried in unstructured scientific literature [16].
The application of machine learning (ML) and deep learning represents a transformative shift in computational PPI prediction. Recently, Transformer-based models, inspired by natural language processing (NLP) successes, have emerged as powerful tools for analyzing biological sequences and text [76]. This technical guide explores the integration of these advanced computational techniques to enhance the extraction and prediction of PPIs from scientific literature, framed within the context of leveraging existing PPI database resources.
Biological and Technical Foundations of PPI Prediction
The Role of PPIs in Cellular Systems and Disease
Protein-Protein Interactions can be categorized as stable or transient, homodimeric or heterodimeric, and direct or indirect, each type shaping specific functional characteristics [28]. These interactions are significant not only for understanding complex molecular processes like plant growth and stress responses but are also vital in human diseases [77] [51]. For instance, in neurodegenerative diseases like Alzheimer's, abnormal PPIs lead to protein aggregation in neural tissue [51]. Similarly, mutations in proteins like KRAS can alter interaction affinities, leading to cancer pathogenesis [51].
Conventional PPI Databases: A User's Perspective
A systematic comparison of PPI databases reveals that no single resource provides comprehensive coverage. Studies show that combined results from STRING and UniHI cover approximately 84% of experimentally verified PPIs, while a combination of hPRINT, STRING, and IID retrieves about 94% of the total available interactions [16]. This highlights the necessity of using multiple databases for comprehensive research. The table below summarizes key PPI databases and their primary characteristics.
Table 1: Essential Protein-Protein Interaction Databases
| Database Name | Description | Key Features | URL |
|---|---|---|---|
| STRING | Known and predicted PPIs across species | Extensive coverage; integrates experimental and predicted data | https://string-db.org/ |
| BioGRID | Protein and genetic interactions from various species | Curated molecular interaction data | https://thebiogrid.org/ |
| IntAct | Protein interaction database | Maintained by EBI; provides a freely available analysis tool | https://www.ebi.ac.uk/intact/ |
| MINT | Focuses on PPIs from high-throughput experiments | Specialized in experimentally verified interactions | https://mint.bio.uniroma2.it/ |
| DIP | Database of experimentally verified PPIs | Catalogs experimentally determined interactions | https://dip.doe-mbi.ucla.edu/ |
| HPRD | Human Protein Reference Database | Includes interaction, enzymatic, and localization data | http://www.hprd.org/ |
| APID | Agile Protein Interaction DataAnalyzer | Offers tools for visualization and analysis | http://apid.dep.usal.es/ |
Machine Learning and Deep Learning Architectures for PPI Prediction
Evolution of Computational Approaches
Early computational methods for PPI prediction relied on manually engineered features, such as sequence similarity, structural alignment, and genomic information [28] [51]. These have been largely superseded by machine learning models, including Support Vector Machines (SVMs) and Random Forests, which improved prediction accuracy by learning from labeled datasets [28]. The subsequent rise of deep learning has brought about a paradigm shift, enabling automatic feature extraction from raw protein sequences and structures, thus capturing complex, non-linear relationships that were previously intractable [28].
Core Deep Learning Models
Modern deep learning architectures for PPI prediction include Convolutional Neural Networks (CNNs), Recurrent Neural Networks (RNNs), and Deep Neural Networks (DNNs) [78]. These models excel at processing high-dimensional biological data and automatically extracting meaningful features [28].
Graph Neural Networks (GNNs) are particularly suited for PPI prediction because they can naturally represent proteins as nodes in a graph, with edges representing interactions or similarities. Key GNN variants include:
- Graph Convolutional Networks (GCNs): Aggregate information from neighboring nodes using convolutional operations [28].
- Graph Attention Networks (GATs): Incorporate an attention mechanism to adaptively weight the importance of neighboring nodes, enhancing flexibility [28].
- GraphSAGE: Designed for large-scale graphs, it uses neighbor sampling and feature aggregation to reduce computational complexity [28].
- Graph Autoencoders (GAEs): Employ an encoder-decoder structure to learn compact node embeddings for tasks like link prediction [28].
Innovative frameworks like AG-GATCN integrate GATs and Temporal Convolutional Networks for robustness against noise, while RGCNPPIS combines GCN and GraphSAGE to extract both macro-scale topological patterns and micro-scale structural motifs [28].
The Transformer Revolution
Transformer-based models have revolutionized NLP and are now making significant inroads into bioinformatics due to their ability to process sequential data, like protein sequences, in parallel and capture long-range dependencies [76].
The core innovation of the Transformer is the self-attention mechanism, which allows the model to weigh the importance of different parts of the input sequence when making predictions. A Transformer model consists of an encoder and a decoder, each comprising stacked layers of multi-head self-attention and position-wise feed-forward networks [76]. For protein sequences, amino acids are treated as tokens, and their embeddings are combined with positional encodings to form the input to the model [76].
Notable Transformer-based protein language models (pLMs) include:
- ProtBERT and ProteinBERT: Used for protein sequence function and structure prediction [76].
- ESM models: A family of models developed by Meta AI for protein structure and function prediction [76].
- TAPE: A framework designed for protein sequence classification and prediction tasks [76].
These pLMs can be fine-tuned for specific downstream tasks, such as predicting whether two protein sequences interact, achieving state-of-the-art performance [76] [51].
Integrated Workflow for PPI Extraction from Literature
This section outlines a comprehensive methodology for leveraging ML and Transformers to extract PPIs from scientific text and sequence data.
System Architecture and Workflow
The following diagram illustrates the integrated workflow for literature-based PPI extraction and prediction, combining text mining and sequence analysis.
Experimental Protocol for PPI Prediction
This protocol details the process for training and evaluating a sequence-based PPI prediction model using deep learning.
Step 1: Data Curation and Preprocessing
- Source Raw Data: Download known PPIs from multiple databases (e.g., STRING, BioGRID, DIP) to create a positive dataset [16]. Generate negative samples by randomly pairing proteins from different cellular compartments or using confirmed non-interacting pairs if available [51].
- Sequence Retrieval: For each protein in the dataset, obtain its amino acid sequence from a dedicated database like UniProt.
- Data Splitting: Partition the protein pairs into training (70%), validation (15%), and test (15%) sets. Implement strict cross-validation at the protein level, ensuring that all pairs involving a specific protein are confined to a single split to prevent data leakage and over-optimistic performance estimates [51].
Step 2: Feature Extraction using Protein Language Models
- Model Selection: Choose a pre-trained pLM such as ESM-2 or ProtBERT [76].
- Generate Embeddings: Pass each protein sequence through the pLM. Extract the embedding from the [CLS] token or compute a mean pooling over all amino acid token embeddings to obtain a fixed-dimensional vector representation for each protein [76].
- Represent Protein Pairs: For a protein pair (A, B), combine their individual embeddings (e.g., by concatenation, element-wise multiplication, or absolute difference) to create a unified feature vector representing the potential interaction [51].
Step 3: Model Training and Fine-Tuning
- Architecture Choice:
- Option A (Classifier): Build a classifier (e.g., a DNN) on top of the combined protein pair embeddings.
- Option B (End-to-End Fine-tuning): For Transformer architectures, add a classification head and fine-tune the entire model on the PPI prediction task, allowing the sequence representations to adapt specifically to interaction data [76].
- Handling Class Imbalance: If negative samples dominate, employ techniques like oversampling the minority class, undersampling the majority class, or using a weighted loss function during training [51].
- Hyperparameter Optimization: Use the validation set to tune hyperparameters (e.g., learning rate, batch size, network depth) to maximize performance metrics.
Step 4: Model Evaluation and Validation
- Performance Metrics: Evaluate the model on the held-out test set using standard metrics [51]. The table below outlines their interpretation.
Table 2: Key Metrics for Evaluating PPI Prediction Models
| Metric | Description | Interpretation in PPI Context |
|---|---|---|
| Accuracy | (TP+TN) / (TP+TN+FP+FN) | Overall correctness, can be misleading if data is imbalanced. |
| Precision | TP / (TP+FP) | The proportion of predicted interactions that are correct. |
| Recall (Sensitivity) | TP / (TP+FN) | The ability to find all true interactions in the data. |
| F1-Score | 2 * (Precision*Recall)/(Precision+Recall) | Harmonic mean of precision and recall. |
| AUC-ROC | Area Under the ROC Curve | Overall model performance across all classification thresholds. |
- Biological Validation: Perform downstream analysis, such as Gene Ontology (GO) term enrichment or pathway analysis (using resources like Reactome), to assess whether predicted interactions are biologically plausible [28]. The most confident novel predictions can be prioritized for experimental validation.
Successfully implementing a PPI extraction pipeline requires a suite of computational tools and data resources.
Table 3: Essential Toolkit for ML-Based PPI Research
| Category | Item/Resource | Function and Application |
|---|---|---|
| Data Resources | STRING, BioGRID, IntAct | Source of ground-truth PPI data for model training and benchmarking [16]. |
| UniProtKB | Primary source for obtaining protein sequences from accessions/names. | |
| Software & Libraries | Python (PyTorch/TensorFlow) | Core programming environment for building and training deep learning models. |
| Hugging Face Transformers | Library providing easy access to pre-trained Transformer models (e.g., ProtBERT). | |
| Biopython | Toolkit for computational biology, useful for sequence manipulation. | |
| Scikit-learn | Library for data preprocessing, model evaluation, and traditional ML. | |
| Computational Models | ESM-2/ESMFold (Meta AI) | State-of-the-art protein language model for generating sequence embeddings [51]. |
| ProtBERT | Transformer model specifically pre-trained on protein sequences [76]. | |
| AlphaFold2/3 | Although structure-based, can provide structural insights to validate or inform predictions [51]. |
Challenges and Future Directions
Despite significant progress, several challenges remain in the application of ML for PPI extraction.
Data Quality and Bias: PPI datasets are often skewed towards well-studied proteins and model organisms, which can limit model generalizability [76] [51]. Noisy annotations and data leakage during training are persistent issues that require careful dataset curation [51].
Model Interpretability: Deep learning models are often considered "black boxes." Developing methods to interpret predictions—for instance, identifying which amino acids or domains are critical for an interaction—is crucial for building trust and gaining biological insights [78].
Integration of Multi-Modal Data: The future lies in multi-omics integration. Combining sequence information with data on gene expression, protein structures, and functional annotations (Gene Ontology) will provide a more holistic view and improve predictive accuracy [77] [28].
Generalizability and Cross-Species Prediction: A key frontier is enhancing model capability to accurately predict interactions for understudied proteins and across different species, which is vital for applications in plant biology (e.g., rice crop improvement) and infectious disease research [77] [51].
In conclusion, the integration of machine learning, particularly Transformer-based models, with established PPI database resources creates a powerful paradigm for accelerating the discovery of protein interactions. This synergy between computational prediction and experimental biology holds immense promise for advancing fundamental biological knowledge and streamlining drug discovery pipelines.
Ensuring Data Quality: A Framework for Benchmarking, Scoring, and Comparative Analysis
Protein-protein interaction (PPI) networks provide a fundamental framework for understanding cellular organization, processes, and functions at a systems level [79]. The analysis of these networks enables researchers to move beyond studying individual proteins to understanding how their collective behavior drives biological phenomena. For researchers and drug development professionals utilizing curated PPI databases such as BioGRID and IntAct, a critical challenge lies in extracting biologically meaningful insights from the vast interaction data. This guide focuses on two pivotal analytical approaches—modularity analysis and enrichment analysis—that transform raw network data into validated biological knowledge. These methods are essential for identifying functionally relevant subnetworks, elucidating disease mechanisms, and pinpointing potential therapeutic targets.
Modularity, a fundamental organizational principle in biological systems, refers to the property of networks to be decomposed into subgroups (modules or communities) characterized by dense internal connections and sparser connections between them [80] [81]. These modules often correspond to protein complexes, functional units, or coordinated pathways. Enrichment analysis provides the statistical foundation for validating the biological significance of these modules by determining whether certain biological annotations (e.g., Gene Ontology terms, pathways) occur within a module more frequently than would be expected by chance. Together, these methods form a powerful validation pipeline for PPI network research.
Core Concepts and Biological Significance
Defining Modularity in Biological Networks
A module in a PPI network is generally defined as a "tightly interconnected set of edges" where "the density of connections inside any so-called module must be significantly higher than the density of connections with other modules" [80] [81]. This structural property is not merely a topological curiosity; it reflects the functional organization of the cell. Functional modules may represent:
- Protein complexes: Groups of proteins that physically assemble to perform a coordinated function [79]
- Signaling pathways: Cascades of interacting proteins that relay cellular signals [81]
- Regulatory units: Sets of proteins involved in the same cellular process or regulated by the same factors [80]
The emergence and preservation of modularity across biological systems is thought to confer significant evolutionary advantages, including robustness (the ability to withstand perturbations), adaptability to changing environments, and functional optimization [80] [81]. From a practical research perspective, identifying modules simplifies the analysis of complex networks by breaking them down into manageable, functionally coherent units.
The Role of Enrichment Analysis in Biological Validation
While modularity analysis identifies candidate functional units, enrichment analysis provides the statistical framework for their biological validation. This method tests whether a set of proteins (e.g., a network module) is statistically enriched with proteins sharing specific biological annotations compared to what would be expected in a random set of proteins of the same size. Common annotation systems used in enrichment analysis include:
- Gene Ontology (GO): Terms describing molecular functions, biological processes, and cellular components
- KEGG Pathways: Curated representations of metabolic and signaling pathways
- Disease Ontology: Associations between genes and human diseases
- Protein Domains: Structural and functional protein domains
A significant enrichment p-value (typically after multiple testing correction) indicates that the observed concentration of a particular annotation in the module is unlikely to be random, thus providing objective evidence of the module's biological relevance [82].
Methodological Framework
Computational Pipeline for Modularity Analysis
A robust workflow for modularity analysis and validation integrates data from PPI databases with advanced analytical algorithms, followed by systematic biological interpretation.
Workflow Diagram: Modularity Analysis Pipeline
Data Acquisition from PPI Databases
The foundation of any network analysis is high-quality interaction data. Key databases provide complementary types of data:
- BioGRID: A curated database of physical and genetic interactions manually extracted from the biomedical literature [2]. As of 2020, it contained over 1.93 million curated interactions from more than 63,000 publications, with specific themed curation projects focusing on areas like the ubiquitin-proteasome system and SARS-CoV-2 interactions [2].
- STRING: A comprehensive resource that integrates both physical and functional associations from experimental data, computational predictions, and prior knowledge [23]. The latest versions incorporate directional regulation information and provide network clustering capabilities.
- DIP: The Database of Interacting Proteins contains experimentally determined protein interactions, often used as a benchmark for method development [79].
Table 1: Key Protein-Protein Interaction Databases for Network Analysis
| Database | Primary Content | Curation Method | Key Features | Use Cases |
|---|---|---|---|---|
| BioGRID [2] | Physical and genetic interactions | Manual curation from literature | High-quality experimental data; themed projects; post-translational modifications | Gold-standard validation; focused pathway analysis |
| STRING [23] | Physical and functional associations | Integration of multiple sources | Comprehensive coverage; confidence scores; pathway enrichment tools | Exploratory analysis; network clustering |
| DIP [79] | Experimentally determined PPIs | Manual and computational curation | Non-redundant dataset; quality evaluation tools | Method development and benchmarking |
Module Detection Algorithms
Several clustering algorithms have been developed specifically for identifying modules in biological networks:
- Markov Cluster Algorithm (MCL): This algorithm "was both remarkably robust to graph alterations and superior for the extraction of complexes from interaction networks" according to a systematic evaluation cited in [79]. MCL simulates random walks on the network graph, using an inflation parameter to control cluster granularity.
- MCODE: Molecular Complex Detection is designed specifically to identify densely connected regions in PPI networks and is available as a Cytoscape app [83].
- Other Approaches: Alternative methods include the Girvan-Newman algorithm, which progressively removes edges with high betweenness centrality, and Louvain method for community detection [80].
In a practical implementation focusing on a pathogen, researchers used MCL with an inflation coefficient of I = 1.8, successfully identifying 172 modules from predicted O157:H7 PPIs, 121 of which were considered highly reliable after evaluation [79].
Enrichment Analysis Methodology
Enrichment analysis statistically evaluates whether certain biological terms occur more frequently in a protein set than expected by chance. The standard approach involves:
- Term Mapping: All proteins in the module are mapped to their associated biological annotations (GO terms, pathway memberships, etc.)
- Statistical Testing: For each term, a statistical test (typically Fisher's exact test) calculates the probability of observing at least as many proteins annotated with that term in the module under the null hypothesis of random distribution
- Multiple Testing Correction: Benjamini-Hochberg procedure or similar methods control the false discovery rate across all tested terms
- Interpretation: Significant terms reveal the module's potential biological functions
Tools like BiNGO, ClueGO, and EnrichmentMap implement these methods within the Cytoscape environment [83].
Experimental Protocols and Validation
Detailed Protocol for Modularity Analysis
Based on established methodologies in the literature [79], here is a detailed protocol for conducting modularity analysis:
Step 1: PPI Network Construction
- Retrieve protein sequences for the organism of interest from RefSeq or similar databases
- Scan proteins with InterProScan to identify domains
- Predict interactions using integrated methods like Maximum Likelihood Estimation (MLE) and Maximum Specificity Set Cover (MSSC) based on known domain-domain interactions
- Apply post-processing to eliminate directional repeats and self-interactions
Step 2: Module Detection with MCL Algorithm
- Format the PPI network as a symmetric adjacency matrix
- Run the MCL algorithm with an inflation parameter typically between 1.8-2.2 (adjust based on desired cluster granularity)
- Process output to identify proteins shared between modules by scanning proteins in each donor cluster for interaction partners in other clusters
Step 3: Quality Evaluation of Predicted Modules
- Compare predicted modules with known protein complexes in databases like CORUM
- Assess functional homogeneity using Gene Ontology annotation consistency
- Validate with pathway databases (KEGG, Reactome)
- Estimate reliability by overlap with known interactions in databases like STRING (approximately 20% overlap is considered good for predicted interactions) [79]
Step 4: Biological Analysis of Modules
- Identify modules containing proteins of interest (e.g., disease-related proteins)
- Visualize intramodular interactions using tools like Cytoscape or Pajek
- Investigate intermodular relationships to understand cross-talk between functional units
Case Study: Modularity Analysis of Root Development in Rice
A recent study demonstrates the practical application of these methods [82]. Researchers extracted the root development module from a global rice PPI network obtained from STRING. Their analysis identified:
- 75 novel candidate proteins involved in root development
- 6 distinct sub-modules representing different functional units
- 20 intramodular hub proteins and 2 intermodular hubs
This systematic approach enabled the researchers to propose new protein candidates and organizational principles for root development, demonstrating how modularity analysis can generate testable biological hypotheses for complex phenotypes.
Research Reagent Solutions
Successful implementation of modularity and enrichment analysis requires a suite of computational tools and databases. The following table summarizes essential resources for researchers.
Table 2: Essential Computational Tools for Network Validation
| Tool/Resource | Type | Primary Function | Application in Analysis |
|---|---|---|---|
| Cytoscape [83] | Desktop Application | Network visualization and analysis | Core platform for network integration and visualization; supports numerous analysis apps |
| MCODE [83] | Cytoscape App | Module detection | Identifies densely connected regions in PPI networks |
| clusterMaker2 [83] | Cytoscape App | Cluster analysis | Provides multiple clustering algorithms for network module detection |
| BiNGO/ClueGO [83] | Cytoscape App | Enrichment analysis | Performs statistical enrichment analysis for GO terms and pathways |
| igraph/NetworkX [83] | Programming Library | Network analysis | Script-based analysis suitable for large networks and pipeline integration |
| Gephi [83] | Desktop Application | Network visualization | Handles large-scale networks with advanced layout algorithms |
| konnect2prot 2.0 [84] | Web Application | Context-specific PPI networks | Generates directional PPI networks with integrated expression analysis |
Implementation Considerations
When establishing a workflow for modularity and enrichment analysis, several practical factors must be considered:
- Computational Resources: The computational load for predicting PPIs and protein modules can be significant, requiring one to two weeks depending on computer power and network size [79]. Large networks may require programmatic solutions like igraph or NetworkX instead of desktop tools [83].
- Tool Selection: For exploratory biological analysis, Cytoscape with specialized apps provides a user-friendly environment. For large-scale analyses or integration into automated pipelines, programmatic solutions like R's igraph or Python's NetworkX are more suitable [83].
- Validation Strategy: A multi-faceted validation approach is essential, incorporating topological metrics, functional enrichment, and comparison with known complexes or pathways.
Advanced Analytical Framework
Integrated Validation Workflow
A comprehensive validation framework for PPI networks combines both topological and biological measures to assess network quality and functional relevance.
Workflow Diagram: Network Validation Framework
Emerging Directions and Future Developments
The field of network biology continues to evolve with several promising developments:
- Directional Regulatory Networks: New capabilities in databases like STRING now incorporate "evidence on the type and directionality of interactions using curated pathway databases and a fine-tuned language model parsing the literature" [23], enabling more precise modeling of regulatory relationships.
- Multi-omics Integration: Tools like konnect2prot 2.0 are bridging "the gap between gene-level regulation and protein-level activity, providing a holistic view of how transcriptional changes fuel cellular behavior" [84] by integrating differential gene expression analysis with PPI networks.
- Machine Learning Applications: The availability of "downloadable network embeddings" from resources like STRING "facilitate the use of STRING networks in machine learning and allow cross-species transfer of protein information" [23].
- CRISPR Integration: Resources like BioGRID-ORCS (Open Repository of CRISPR Screens) capture genetic interaction data from CRISPR/Cas9 screens, enabling integration of functional genetic data with physical interaction networks [2].
These advancements are creating increasingly sophisticated frameworks for validating PPI networks and extracting biologically meaningful insights with potential applications in drug discovery and therapeutic target identification.
The systematic mapping of protein-protein interactions (PPIs) has become a fundamental aspect of systems biology, providing crucial insights into cellular organization and function. However, high-throughput experimental methods for detecting PPIs, such as yeast two-hybrid (Y2H) screens and affinity purification followed by mass spectrometry (AP/MS), are known to yield substantial rates of false positives and false negatives [85] [86]. Consequently, confidence scoring systems have emerged as essential tools to distinguish biologically relevant interactions from spurious data, thereby enabling researchers to construct more reliable interaction networks for downstream analysis [87]. The primary goal of these scoring systems is to assign a quantitative measure—typically a probability or a score between 0 and 1—that reflects the likelihood that a reported interaction represents a true biophysical interaction within the cell [88].
The need for such standardized assessment is particularly critical for large-scale projects, such as the human interactome project, where standardized experimental methods for quality control allow the scientific community to evaluate data under a universally interpretable quality standard [85]. Confidence scores do not indicate the strength or specificity of an interaction but rather serve as indicators of reliability, helping researchers filter datasets and prioritize interactions for experimental validation [88].
Major Methodological Approaches for Confidence Assessment
Experimental Validation and Benchmarking
The most direct approach for assessing interaction reliability involves experimental validation using complementary assays. A pioneering methodology developed a standardized confidence-scoring method based on a tool-kit of four complementary high-throughput protein interaction assays [85]. These assays were systematically benchmarked against defined reference sets:
- Positive Reference Set (PRS): Consists of well-documented human interaction pairs supported by multiple peer-reviewed publications and manually curated across multiple databases.
- Random Reference Set (RRS): Comprises randomly chosen protein pairs from the human ORFeome, serving as a negative control set since randomly chosen pairs are unlikely a priori to interact [85].
The core experimental workflow involves testing all pairs from these reference sets across the assay tool-kit. The resulting data is used to train a logistic regression model, which calculates the probability that any novel interaction pair is a true biophysical interaction once tested in the tool-kit [85]. The workflow for this experimental validation approach is detailed in Figure 1.
Figure 1. Workflow for Experimental Validation of PPIs. The process begins with a PPI dataset and tests it against positive and random reference sets using complementary assays. Results are benchmarked and used to train a model that calculates final confidence scores. (Adapted from [85])
Table 1: Performance of Individual Assays on Reference Sets (hsPRS-v1 and hsRRS-v1)
| Assay Method | Principle | Assay Sensitivity (% of PRS Detected) | False Positives (RRS Detected) |
|---|---|---|---|
| LUMIER | Pull-down assay with luciferase readout | 36% | 4 pairs |
| MAPPIT | Ligand-dependent luciferase readout in mammalian cells | 33% | 2 pairs |
| Y2H | Transcription-based in yeast | 25% | 0 pairs |
| PCA | Protein fragment complementation | 23% | 2 pairs |
| wNAPPA | Completely in vitro protein array | 21% | 3 pairs |
The performance characteristics reveal that while LUMIER showed the highest sensitivity, it also detected several false positives. All methods demonstrated modest sensitivity (20-35%), underscoring the necessity of using orthogonal approaches rather than relying on a single assay [85].
Computational and Topology-Based Scoring
Computational methods leverage the structure of interaction networks themselves to assess reliability, requiring no additional experimental data.
- The IRAP Measure: The "interaction reliability by alternative path" (IRAP) method operates on the principle that a candidate PPI is considered reliable if it is involved in a closed loop where the alternative path of interactions between the two interacting proteins is strong. This global, system-wide approach that considers the entire interaction network has been shown to be more effective than local neighbor methods [89].
- The IDBOS Method: This unsupervised statistical approach scores PPIs by comparing the actual observed occurrence of an interaction in aggregated datasets with its expected occurrence in randomized samples. The method calculates a Z-score, defined as ( Z{ij} = (O{ij} - \langle R{ij} \rangle) / \sigmaR ), where ( O{ij} ) is the observed count and ( \langle R{ij} \rangle ) is the average count from randomizations. This approach corrects for biases toward frequently studied proteins and allows for rank-ordering of PPIs based on statistical significance [87].
- The Semantic Reliability (SR) Method: This approach assesses reliability based on the semantic relationship between protein functions. It combines two measures: the external reliability (semantic influence between proteins that interact with the target proteins) and the internal reliability (semantic relationship between the two target proteins themselves). This method leverages functional similarity and shared biological roles to assess interaction plausibility [86].
Integrated Scoring in Public Databases
Public resources like STRING integrate multiple evidence channels to compute unified confidence scores. STRING combines probabilities from various evidence types—including experimental data, gene co-occurrence, text mining, and transferred evidence from other organisms—while correcting for the probability of randomly observing an interaction [88]. The combined scores range from 0 to 1, with higher scores indicating greater confidence. STRING suggests specific thresholds for different confidence levels: 0.15 (low), 0.40 (medium), 0.70 (high), and 0.90 (highest confidence) [90]. A comparative analysis of database coverage found that combined use of STRING and UniHI retrieved approximately 84% of experimentally verified PPIs, demonstrating the effectiveness of integrated scoring systems [16].
Quantitative Comparison of Scoring Systems
Different scoring methods exhibit varying performance characteristics in terms of their ability to identify biologically relevant interactions. The following table summarizes the enrichment provided by different ranking methods compared to randomly ranked data, based on retrieval of known biological associations:
Table 2: Performance Comparison of PPI Ranking Methods
| Ranking Method | Basis of Scoring | Enrichment over Random Ranking | Key Advantages |
|---|---|---|---|
| IDBOS | Statistical over-representation vs. randomized networks | ~134% | Unsupervised, corrects for study bias, works without external data |
| Hypergeometric Test | Probability of observed co-occurrence | ~109% | Standard statistical foundation |
| Simple Occurrence | Number of literature reports | ~46% | Intuitive, easy to compute |
| IRAP | Network topology (alternative paths) | Better than IG2/IG1 | Global network perspective |
| Semantic Reliability | Functional similarity | Outperforms IG1/IG2 on yeast data | Incorporates biological context |
The IDBOS method demonstrates superior performance by effectively normalizing the bias where proteins that are frequently studied (popular) accumulate a disproportionately high number of reported interactions, which may not reflect biological reality [87].
Practical Considerations for Researchers
Threshold Selection and Robustness in Network Analysis
Choosing an appropriate confidence threshold is a critical practical decision when working with PPI data. Setting the threshold too high may result in excess false negatives, while setting it too low increases false positives. Research has shown that the choice of score threshold can significantly affect network topology metrics, such as average degree and clustering coefficient, which in turn impacts the identification of key proteins [90].
To address this, robustness measures—rank continuity, identifiability, and instability—have been developed to evaluate how consistent node metrics (e.g., centrality measures) are across different thresholds. Studies evaluating 25 node metrics found that the number of edges in the step-1 ego network, along with leave-one-out difference (LOUD) metrics for average redundancy and natural connectivity, were significantly more robust to threshold choice than commonly used metrics like betweenness centrality [90]. The logical relationship between threshold choice and analytical outcomes can be visualized as a decision process, as shown in Figure 2.
Figure 2. Threshold Selection and Robustness Assessment. Selecting a confidence threshold involves trade-offs between network coverage and data quality. Assessing the robustness of findings across thresholds strengthens biological conclusions. (Adapted from [90])
Research Reagent Solutions for Experimental Validation
For researchers seeking to experimentally validate PPIs, specific reagent solutions and experimental systems have been benchmarked for performance. The following table details key research reagents and their applications in PPI confidence assessment:
Table 3: Key Research Reagent Solutions for PPI Validation
| Reagent/Resource | Type | Function in Validation | Key Characteristics |
|---|---|---|---|
| Yeast Two-Hybrid (Y2H) Systems | Biological Assay | Detects binary interactions in yeast nuclei | Uses transcriptional activation; multiple strain options (e.g., Y8800, MaV103) affect sensitivity [85] |
| MAPPIT | Biological Assay | Detects interactions in mammalian cells | Ligand-dependent luciferase readout; more physiologically relevant PTM context [85] |
| LUMIER | Biochemical Assay | Pull-down with luciferase reporter | High sensitivity (36%); detects phosphorylation-dependent interactions [85] |
| Protein Complementation (PCA) | Biological Assay | Fluorescent protein reconstitution in cells | Strong, irreversible signals; requires careful optimization to minimize false positives [85] |
| wNAPPA | In Vitro Assay | Protein array with in vitro transcription/translation | Cell-free system; 21% sensitivity on benchmark sets [85] |
| STRING Database | Computational Resource | Provides pre-computed confidence scores | Integrates multiple evidence channels; suggests confidence thresholds [88] [90] |
| Positive Reference Set (PRS) | Reference Material | Gold standard for true interactions | Contains well-documented human protein pairs; essential for benchmarking [85] |
Confidence scoring systems are indispensable for leveraging high-throughput PPI data in biological research and drug development. The integration of orthogonal approaches—experimental validation, computational scoring, and functional annotation—provides the most robust framework for identifying reliable interactions. As interaction databases continue to grow and evolve, the development of more sophisticated scoring methodologies that account for network dynamics, contextual information, and multi-optic data integration will further enhance our ability to distinguish true biological interactions from experimental noise, ultimately advancing our understanding of cellular systems and facilitating drug target identification.
The systematic study of Protein-Protein Interaction (PPI) networks is a cornerstone of modern systems biology, providing critical insights into cellular signaling, functional annotation, and disease mechanisms. For researchers, scientists, and drug development professionals, the construction of these networks relies heavily on data extracted from curated public databases. However, the presence of spurious interactions and the fragmented nature of the data landscape present significant challenges. Different databases often report varying numbers of interactions for the same publication due to differences in curation practices, identifier mapping, and data representation models [24]. This technical guide provides an in-depth framework for the comparative analysis of integrated PPI networks, with a specific focus on assessing their performance under different integration stringencies. Framed within the broader context of utilizing resources like IntAct and BioGRID, this work emphasizes practical methodologies for data integration, quality assessment, and robust network construction to support high-confidence biological discovery.
The Landscape of Protein-Protein Interaction Databases
A comprehensive understanding of the available PPI resources is the first step in any network analysis. The field is characterized by a large number of databases that differ significantly in scope, content, and curation philosophy.
Key Public PPI Databases and Their Characteristics
Historically, six major databases have been primary repositories for experimentally verified PPIs: the Biological General Repository for Interaction Datasets (BioGRID), the Molecular INTeraction database (MINT), the Biomolecular Interaction Network Database (BIND), the Database of Interacting Proteins (DIP), the IntAct molecular interaction database (IntAct), and the Human Protein Reference Database (HPRD) [24]. These databases collectively curate interactions from thousands of scientific publications, yet their coverage for any specific organism or study can vary dramatically. For instance, while IntAct might report the highest number of unique interactions from high-throughput studies, HPRD often provides more comprehensive coverage from small-scale publications, particularly for human proteins [24].
To address the challenge of data fragmentation, meta-databases and integration tools have been developed. The International Molecular Exchange (IMEx) consortium, with members including IntAct, MINT, and DIP, aims to enable data exchange and avoid duplication of curation efforts using a standardized data format (PSI-MI) [24] [91]. Furthermore, tools like the Protein Interaction Network Online Tool (PINOT) automate the process of querying, downloading, and integrating PPI data from multiple IMEx-associated repositories and WormBase in real-time, applying user-defined quality filters [91].
Quantitative Comparison of Database Coverage
A systematic comparison of 16 human PPI databases revealed critical differences in their coverage of experimentally verified and predicted interactions. The analysis, which used a query set of 108 genes associated with specific tissues and diseases, found that a combined dataset from STRING and UniHI could retrieve approximately 84% of all 'experimentally verified' PPIs available across the studied resources [16]. For a more complete picture that includes both experimental and predicted interactions, the combined use of hPRINT, STRING, and IID was necessary, recovering about 94% of the 'total' available PPIs [16]. Notably, among the exclusively found experimentally verified PPIs, STRING contributed around 71% of these unique hits, underscoring its value in a comprehensive integration strategy [16].
Table 1: Coverage of Protein-Protein Interactions Across Major Databases
| Database | Primary Focus | Reported Interactions (2008 Data) | Notable Strengths |
|---|---|---|---|
| IntAct | High-throughput studies | ~129,559 (from 131 organisms) | Largest number of unique interactions; IMEx member [24]. |
| BioGRID | Genetic & protein interactions | ~90,972 (from 10 organisms) | Cites a high number of publications (~16,369) [24]. |
| HPRD | Human proteins | ~36,169 (human only) | Comprehensive coverage from over 18,000 publications; includes other protein data [24]. |
| MINT | Molecular interactions | ~80,039 (from 144 organisms) | IMEx member [24]. |
| STRING | Experimental & predicted | N/A (Wide coverage) | Covers ~71% of exclusive experimentally verified PPIs; combines data from many sources [16]. |
Methodologies for Integration and Assessment
Constructing a unified PPI network from multiple sources requires a defined experimental protocol. The following workflow outlines the key steps, from data acquisition to final assessment, and can be adapted for various research questions.
Experimental Protocol for Network Integration
The process begins with the acquisition of raw PPI data. This can be achieved by downloading complete datasets from individual database websites or, more efficiently, by using a programmatic tool like PINOT, which queries multiple sources simultaneously via the PSICQUIC interface [91]. The initial query should consist of a list of proteins of interest (seed proteins) in an approved format, such as HGNC gene symbols or UniProt IDs for human proteins.
Once collected, the data must be parsed and merged. This involves mapping protein identifiers to a consistent namespace (e.g., UniProt ID) to resolve conflicts arising from different naming conventions used by the source databases. This step is critical, as identifier mapping issues are a common source of data loss or inaccuracy during integration [24].
The core of the methodology is the application of integration stringency filters. These filters are used to control the quality and quantity of interactions included in the final network.
- Lenient Integration: This approach aims for maximum coverage. It includes all reported binary physical interactions, regardless of the number of supporting publications or detection methods. This is useful for exploratory analyses but carries a higher risk of including false positives.
- Stringent Integration: This conservative approach applies confidence thresholds to build a high-confidence network. A common and transparent method, as implemented in PINOT, is to score each interaction based on two criteria [91]:
- Method.Score: The number of distinct experimental methods (e.g., Y2H, TAP-MS) used to detect the interaction.
- Publication.Score: The number of different peer-reviewed publications that report the interaction. A Final.Score is calculated (e.g., Method.Score + Publication.Score), and a threshold is applied. For example, a Final.Score of 2 or higher would require an interaction to be reported by at least two independent publications, by two different methods, or by one publication using two methods [91].
Finally, the integrated and filtered network is output in a standard format (e.g., a tab-delimited file) that can be directly used for downstream analysis and visualization in network analysis software.
Workflow Visualization
The diagram below illustrates the logical workflow for the integration and assessment of PPI networks at different stringencies.
Performance Assessment Metrics
Evaluating the performance of integrated networks constructed under different stringencies is essential for interpreting subsequent biological findings. Assessment should focus on both topological characteristics and functional coherence.
Topological and Functional Metrics
- Network Density and Connectivity: A lenient network will typically be larger and denser, potentially revealing a more extensive web of connections. However, a stringent network, while sparser, should exhibit a higher proportion of reliable interactions. The preservation of well-established protein complexes and pathways in the stringent network is a key indicator of quality.
- Coverage of a Gold-Standard Set: A robust quantitative method is to measure the network's coverage of a set of literature-curated, experimentally proven PPIs (a "gold-standard" set). Analysis has shown that databases like GPS-Prot, STRING, APID, and HIPPIE can each cover approximately 70% of such curated interactions [16]. The recovery rate of these gold-standard interactions in your integrated network at different stringencies is a direct measure of sensitivity.
- Functional Enrichment Analysis: This assesses the biological relevance of the network. For a set of seed proteins known to be involved in a specific pathway or disease, a high-quality network should show significant functional enrichment for the expected Gene Ontology (GO) terms, biological pathways (e.g., from KEGG or Reactome), and disease associations. A stringent network often yields more statistically significant enrichment results due to a lower noise level.
- Skewed Coverage Analysis: It is important to note that the coverage of certain databases can be skewed for some gene types [16]. Therefore, performance assessment should consider whether the integrated network adequately represents the specific biological context under investigation (e.g., a specific tissue, disease, or functional class of proteins).
Table 2: Performance Characteristics of Network Integration Strategies
| Assessment Metric | Lenient Integration Strategy | Stringent Integration Strategy |
|---|---|---|
| Primary Objective | Maximize interaction coverage [91] | Maximize confidence in interactions [91] |
| Typical Workflow | Combine all PPIs from multiple databases with minimal or no scoring filters. | Apply confidence thresholds based on replication (e.g., methods, publications) [91]. |
| Expected Outcome | Larger, denser network with higher potential for false positives. | Smaller, more sparse network of high-confidence interactions. |
| Best Use-Cases | Exploratory analysis; hypothesis generation; studying poorly characterized proteins. | Pathway validation; drug target prioritization; deriving robust biological conclusions. |
| Key Advantage | Retrieves up to 94% of total available PPIs (via hPRINT, STRING, IID) [16]. | Covers ~70% of gold-standard, curated interactions (e.g., via GPS-Prot, STRING) [16]. |
Success in integrated network analysis depends on leveraging a suite of key databases, software tools, and standards. The following table details essential "research reagents" for the field.
Table 3: Key Resources for Integrated PPI Network Analysis
| Resource Name | Type | Function & Utility |
|---|---|---|
| PSICQUIC [91] | Programmatic Interface | A standardized interface to simultaneously query multiple PPI databases, enabling efficient data acquisition. |
| IMEx Consortium [24] | Data Standardization | A consortium of major databases that adheres to PSI-MI standards to facilitate data exchange and reduce curation overlap. |
| PINOT [91] | Web Tool / R Scripts | Automates the process of querying PSICQUIC, integrating data, and applying confidence scoring based on methods and publications. |
| STRING [16] | Meta-Database | A comprehensive resource combining experimental and predicted PPIs from many sources; crucial for broad coverage. |
| HGNC Symbols [91] | Nomenclature | Approved human gene nomenclature; using this as a standard for queries avoids identifier mapping issues. |
| UniProt ID [91] | Nomenclature | A standardized protein identifier; essential for accurately merging data from different source databases. |
| Gold-Standard PPI Set [16] | Validation Set | A set of literature-curated, experimentally proven PPIs used to benchmark and assess the performance of an integrated network. |
The construction of integrated PPI networks is not a one-size-fits-all process. The choice between a lenient or stringent integration strategy directly shapes the resulting network's properties and should be guided by the specific biological question. Lenient integration maximizes coverage, which is valuable for exploratory research, while stringent integration, which filters interactions based on replicability across methods and publications, yields higher-confidence networks suitable for validation studies and translational research. The existence of significant discrepancies between databases—where the same publication can yield different interaction counts—underscores the non-trivial nature of data integration and the critical need for the methodologies and comparative assessments described in this guide [24]. By systematically applying these protocols and performance metrics, researchers can build more reliable and biologically meaningful networks, thereby strengthening the foundation for discoveries in systems biology and drug development.
Benchmarking Against Gold-Standard Datasets and Experimental Validation
Protein-protein interaction (PPI) research constitutes a foundational element of modern biology, enabling scientists to decipher the complex regulatory mechanisms governing cellular processes, signal transduction, and disease pathways. The exponential growth of PPI data, fueled by high-throughput technologies and computational predictions, has created an urgent need for systematic benchmarking against gold-standard datasets and rigorous experimental validation frameworks. Within the broader ecosystem of PPI data resources such as IntAct and BioGRID, benchmarking serves as the critical quality control mechanism that ensures data reliability, facilitates method comparisons, and ultimately translates computational insights into biologically meaningful findings. This whitepaper provides an in-depth technical examination of contemporary benchmarking methodologies, gold-standard dataset creation, experimental validation protocols, and their collective importance for researchers, scientists, and drug development professionals engaged in PPI research.
The establishment of standardized benchmarks has become increasingly crucial as deep learning approaches revolutionize PPI prediction. These models, including graph neural networks (GNNs), convolutional neural networks (CNNs), and protein language models (PLMs), have demonstrated remarkable capabilities in predicting interactions from sequence and structural data [28]. However, without rigorous benchmarking against experimentally validated gold standards, the performance claims of these models remain unverifiable, potentially compromising their utility in critical applications such as drug target identification. This technical guide addresses precisely this gap by providing a comprehensive framework for benchmarking and validation tailored to the needs of the PPI research community.
Gold-Standard Datasets for PPI Research
Characteristics and Construction of Gold-Standard Datasets
Gold-standard datasets in PPI research represent carefully curated collections of protein interactions with strong experimental evidence supporting their existence. These datasets serve as reference points for training computational models, benchmarking prediction algorithms, and validating new interactions. The construction of these resources involves meticulous curation processes, expert annotation, and stringent quality control measures to ensure data reliability.
A prime example of such effort is the RAGPPI (RAG Benchmark for Protein-Protein Interactions) benchmark, introduced in 2025 to specifically address the challenge of identifying biological impacts of PPIs for target identification in drug development. This comprehensive benchmark comprises 4,420 question-answer pairs focusing on potential biological impacts of PPIs, including a gold-standard dataset of 500 QA pairs developed through expert-driven data annotation and a silver-standard dataset of 3,720 QA pairs constructed using an ensemble auto-evaluation LLM that reflected expert labeling characteristics [92]. The creation of RAGPPI involved interviews with domain experts to establish criteria for benchmark datasets, including question-answer types and source selection, ensuring relevance to real-world research needs.
Table 1: Key Databases for PPI Research
| Database Name | Description | Key Features | URL |
|---|---|---|---|
| BioGRID | Database of protein-protein and genetic interactions | Covers multiple species; 2.25M+ non-redundant interactions; monthly updates | thebiogrid.org |
| IntAct | Protein interaction database | Molecular interaction data; curated by EBI | ebi.ac.uk/intact |
| STRING | Known and predicted protein-protein interactions | Functional associations; multiple evidence channels | string-db.org |
| MINT | Protein-protein interactions | Focus on high-throughput experiments | mint.bio.uniroma2.it |
| HPRD | Human protein reference database | Interaction, enzymatic, and cellular localization data | hprd.org |
| DIP | Experimentally verified protein-protein interactions | Database of Interacting Proteins | dip.doe-mbi.ucla.edu |
BioGRID represents one of the most comprehensive PPI resources, regularly updated with new interactions and currently containing over 2.25 million non-redundant interactions curated from more than 87,000 publications as of November 2025 [22]. The database employs sophisticated evidence coding to categorize interaction types, enabling researchers to filter interactions based on experimental evidence quality and methodology. This granular approach to data annotation makes BioGRID particularly valuable for constructing specialized benchmarking datasets focused on specific experimental methodologies or interaction types.
Specialized Benchmark Datasets for Method Evaluation
Beyond general-purpose PPI databases, specialized benchmark datasets have emerged to address specific computational challenges. The multi-species dataset created by Sledzieski et al.., for instance, has been widely adopted for cross-species PPI prediction benchmarking [93]. This dataset includes human training data with 421,792 protein pairs (38,344 positive interactions and 383,448 negative pairs), with separate validation and test sets for mouse, worm, fly, yeast, and E. coli. The careful construction of this dataset, with positive PPIs derived from experimental evidence and negative pairs representing randomly paired proteins not reported to interact, provides a robust framework for evaluating model generalizability across evolutionary distances.
For hierarchical relationship analysis, datasets such as SHS27K and SHS148K derived from STRING provide Homo sapiens-specific PPI networks with 12,517 and 44,488 interactions respectively [75]. These datasets are particularly valuable for evaluating methods that capture the natural hierarchical organization of PPI networks, ranging from molecular complexes to functional modules and cellular pathways.
Experimental Validation Frameworks
Experimental Evidence Codes and Methodologies
Experimental validation of PPIs relies on diverse methodologies, each with specific strengths, limitations, and appropriate application contexts. BioGRID has established a comprehensive classification system for experimental evidence codes that serves as a valuable framework for designing validation strategies [20].
Table 2: Key Experimental Methods for PPI Validation
| Method Category | Specific Techniques | Key Applications | Evidence Level |
|---|---|---|---|
| Affinity Capture | Affinity Capture-MS, Affinity Capture-Western, Affinity Capture-RNA | Identification of direct binding partners | Direct Physical |
| Biophysical Methods | Co-crystal Structure, FRET, PCA, Surface Display | Structural characterization and proximity detection | Direct Physical |
| Biochemical Assays | Biochemical Activity, Far Western, Protein-peptide | Functional interaction assessment | Direct/Functional |
| Genetic Methods | Two-hybrid, Dosage Growth Defect, Dosage Lethality | Genetic interaction mapping | Genetic |
| Proximity-based | Proximity Label-MS, Cross-Linking-MS | In vivo interaction mapping | Direct Physical |
Physical Interaction Methods include techniques such as Affinity Capture-Mass Spectrometry (Affinity Capture-MS), where a bait protein is affinity-captured from cell extracts and associated interaction partners are identified by mass spectrometric methods [20]. This approach provides evidence of direct physical binding under near-physiological conditions. Similarly, Co-crystal Structure analysis demonstrates interactions at atomic resolution through X-ray crystallography, NMR, or Electron Microscopy, offering unparalleled structural insights but requiring protein crystallization.
Functional Interaction Methods include techniques such as Biochemical Activity assays, which infer interaction from the biochemical effect of one protein upon another in vitro, such as phosphorylation of a substrate by a kinase [20]. These methods establish functional relationships between proteins but may not always demonstrate direct physical contact. The Two-hybrid system infers interactions through reporter gene activation when bait and prey proteins interact, enabling high-throughput screening but potentially yielding higher false-positive rates in some contexts.
Validation Workflows and Experimental Design
A robust experimental validation workflow for PPIs typically follows a multi-stage approach, progressing from initial screening to confirmatory validation. The diagram below illustrates a comprehensive validation workflow integrating multiple experimental methodologies:
Primary Validation typically begins with high-throughput methods such as yeast two-hybrid screening or affinity capture-mass spectrometry, which enable efficient screening of multiple potential interactions. These methods provide initial evidence but may require confirmation through orthogonal approaches due to potential false positives.
Secondary Validation employs orthogonal techniques such as co-immunoprecipitation (Co-IP), fluorescence resonance energy transfer (FRET), or protein-fragment complementation assays (PCA) to confirm interactions detected in primary screens. These methods strengthen evidence by demonstrating interactions through different physical principles or experimental conditions.
Tertiary and Functional Validation provides high-confidence evidence through structural characterization (e.g., X-ray crystallography), detailed biochemical analyses, or assessment of functional consequences through mutational studies or genetic interaction profiling. This tiered approach ensures comprehensive validation while efficiently allocating resources.
Computational Benchmarking Methodologies
Performance Metrics and Evaluation Frameworks
Rigorous benchmarking of PPI prediction methods requires standardized performance metrics and evaluation protocols. The area under the precision-recall curve (AUPR) has emerged as a particularly informative metric for PPI prediction due to the typically imbalanced nature of PPI datasets, where non-interacting pairs often substantially outnumber interacting pairs [93]. The area under the receiver operating characteristic curve (AUROC) provides complementary information about overall classification performance, while metrics such as F1-score, precision, and recall offer insights into specific aspects of predictive performance.
Cross-species validation represents a particularly stringent benchmarking approach that assesses model generalizability beyond training data. In recent benchmarks, models trained on human PPI data were evaluated on mouse, fly, worm, yeast, and E. coli datasets to test their ability to generalize across evolutionary distances [93]. Performance typically correlates with sequence similarity, with higher performance observed between closely related species (e.g., human-mouse) and more challenging prediction for evolutionarily distant species (e.g., human-yeast).
Benchmarking Results for State-of-the-Art Methods
Recent comprehensive benchmarking studies have evaluated the performance of contemporary PPI prediction methods across multiple datasets and species. The table below summarizes performance metrics for leading methods based on cross-species validation:
Table 3: Performance Comparison of PPI Prediction Methods (AUPR)
| Method | Human | Mouse | Fly | Worm | Yeast | E. coli |
|---|---|---|---|---|---|---|
| PLM-interact | 0.852 | 0.841 | 0.798 | 0.763 | 0.706 | 0.722 |
| TUnA | 0.834 | 0.824 | 0.739 | 0.719 | 0.641 | 0.675 |
| TT3D | 0.721 | 0.725 | 0.660 | 0.636 | 0.553 | 0.605 |
| D-SCRIPT | 0.683 | 0.692 | 0.621 | 0.598 | 0.512 | 0.534 |
| PIPR | 0.645 | 0.658 | 0.587 | 0.562 | 0.486 | 0.501 |
| DeepPPI | 0.612 | 0.629 | 0.554 | 0.531 | 0.452 | 0.478 |
PLM-interact, which extends protein language models (ESM-2) through joint encoding of protein pairs and next-sentence prediction fine-tuning, has demonstrated state-of-the-art performance in cross-species benchmarks [93]. This approach achieves significant improvements over previous methods, with AUPR increases of 2-10% compared to the next best method (TUnA) across different species. The performance advantage stems from its ability to directly model inter-protein relationships rather than relying solely on intra-protein features extracted from single sequences.
For hierarchical PPI network analysis, HI-PPI has shown superior performance, improving Micro-F1 scores by 2.62%-7.09% over the second-best method on standard benchmarks [75]. This framework integrates hyperbolic graph convolutional networks with interaction-specific learning to capture both the hierarchical organization of PPI networks and unique interaction patterns between specific protein pairs.
Successful PPI research requires access to specialized reagents, computational tools, and data resources. The following table catalogues essential components of the PPI researcher's toolkit:
Table 4: Essential Research Reagents and Resources for PPI Studies
| Resource Category | Specific Resources | Application/Function |
|---|---|---|
| Experimental Systems | Yeast Two-Hybrid System, Affinity Capture Tags (GST, His, etc.), Cross-linking Reagents | Detection of direct physical interactions between proteins |
| Detection Reagents | Specific Antibodies, Fluorescent Protein Tags (CFP, YFP), Proximity Labeling Enzymes (BioID) | Visualization and quantification of protein interactions |
| Computational Tools | PLM-interact, HI-PPI, TUnA, TT3D, D-SCRIPT | Prediction and analysis of PPIs from sequence and structural data |
| Data Resources | BioGRID, IntAct, STRING, PDB, CORUM | Access to curated interaction data and protein information |
| Validation Resources | Mutagenesis Kits, CRISPR-Cas9 Systems, Recombinant Protein Expression Systems | Experimental validation of interaction interfaces and functional consequences |
Protein Language Models such as ESM-2 have become indispensable tools for representing protein sequences in computational predictions. These models, pre-trained on millions of protein sequences, capture evolutionary information, structural features, and functional constraints that inform interaction potential [93]. Fine-tuning strategies, such as the next-sentence prediction approach used in PLM-interact, adapt these general-purpose models to specifically recognize interaction patterns between protein pairs.
Graph Neural Networks including Graph Convolutional Networks (GCNs), Graph Attention Networks (GATs), and GraphSAGE provide powerful frameworks for analyzing PPI networks [28]. These architectures naturally represent proteins as nodes and interactions as edges, enabling learning from both node features and network topology. Recent innovations such as hyperbolic GCNs further enhance these models' ability to capture the hierarchical organization inherent in biological systems [75].
Integrated Workflow for Benchmarking and Validation
A comprehensive approach to PPI benchmarking and validation integrates computational and experimental methodologies throughout the research lifecycle. The following diagram illustrates this integrated workflow:
This integrated workflow emphasizes the cyclical nature of PPI research, where computational predictions inform experimental designs, experimental results validate and refine computational models, and newly validated interactions expand gold-standard datasets to enable more robust benchmarking in future iterations. Implementation of this framework requires close collaboration between computational and experimental researchers, shared data standards, and adherence to community-established benchmarking protocols.
Benchmarking against gold-standard datasets and experimental validation constitutes an essential discipline within PPI research, ensuring the reliability and biological relevance of computational predictions. As deep learning approaches continue to advance, incorporating increasingly sophisticated architectures from natural language processing and graph representation learning, the importance of rigorous benchmarking and validation only grows more critical. The development of specialized benchmarks such as RAGPPI for biological impact assessment represents a promising direction toward more application-relevant evaluation [92].
Future advancements in PPI benchmarking will likely focus on several key areas: (1) development of more sophisticated negative examples that better reflect biological reality beyond random pairing; (2) integration of temporal and contextual dimensions to account for condition-specific interactions; (3) standardized evaluation frameworks for predicting interaction interfaces and binding affinities; and (4) community-wide adoption of benchmarking protocols to enable direct comparison across studies. Additionally, as protein language models and other AI approaches become more prevalent, there will be increasing need for benchmarks that assess not just predictive accuracy but also biological interpretability, computational efficiency, and utility for drug discovery applications.
For researchers embarking on PPI studies, adherence to the principles and methodologies outlined in this technical guide provides a robust foundation for generating biologically meaningful, computationally rigorous, and experimentally verifiable insights into the complex interactome that underpins cellular function and dysfunction in disease.
Best Practices for Selecting the Optimal Database or Combination for Your Research Goal
In the field of protein-protein interaction (PPI) research, the selection of appropriate databases is a critical foundational step that directly impacts the quality, comprehensiveness, and reliability of research outcomes. The absence of periodic systematic comparisons often forces biologists and bioinformaticians to make subjective selections among the many available PPI databases and tools [16]. With over 375 PPI resources compiled in recent literature and approximately 125 considered important, researchers face a significant challenge in navigating this complex landscape [16]. This technical guide provides evidence-based methodologies for selecting optimal PPI databases and combinations tailored to specific research objectives, experimental designs, and analytical requirements within the context of modern drug development and basic research.
The protein-protein interaction network landscape has expanded dramatically, with databases differing substantially in scope, content, annotation standards, and curation approaches [24]. These resources collect published PPI data and provide researchers access to curated datasets, typically referencing original publications and experimental methods used to determine each interaction. However, database designers choose to represent these data in different ways, and the wide spectrum of experimental methods makes it difficult to design a single data model to capture all necessary experimental detail [24]. This heterogeneity necessitates a systematic approach to database selection based on quantitative metrics and proven methodologies rather than convenience or familiarity alone.
Comparative Analysis of Major PPI Databases
Key Selection Metrics and Performance Indicators
When evaluating PPI databases for research use, both quantitative metrics and qualitative factors must be considered. Quantitative comparisons of database coverage should assess both 'experimentally verified' and 'total' (experimentally verified and predicted) PPIs [16]. Coverage analysis should be performed using gene queries representative of the research focus, as database performance may vary significantly across different gene types and functional categories.
Table 1: Experimental Coverage of Major PPI Databases
| Database | Primary Focus | Interaction Count | Organism Coverage | Key Strengths |
|---|---|---|---|---|
| IntAct | Comprehensive molecular interaction data | ~129,559 interactions [24] | 131 organisms [24] | Extensive high-throughput data; IMEx member |
| BioGRID | Genetic and protein interactions | ~90,972 interactions [24] | 10 organisms [24] | Extensive publication coverage (~16,369) [24] |
| HPRD | Human protein reference | ~36,169 interactions [24] | Human-specific [24] | Comprehensive human data; includes modifications and disease associations |
| MINT | Molecular interactions | ~80,039 interactions [24] | 144 organisms [24] | Focus on curated molecular interactions |
| STRING | Known and predicted interactions | N/A | Extensive | Integration of predicted interactions with experimental data [16] |
| UniHI | Unified human interactome | N/A | Human-focused | Complementary coverage of experimentally verified interactions [16] |
Research comparing the coverage of 16 carefully-selected databases related to human PPIs has demonstrated that combined results from STRING and UniHI covered approximately 84% of 'experimentally verified' PPIs, while about 94% of the 'total' PPIs available across databases were retrieved by the combined use of hPRINT, STRING, and IID [16]. Among the experimentally verified PPIs found exclusively in each database, STRING contributed around 71% of the unique hits [16]. These findings highlight the critical importance of strategic database combinations rather than reliance on single sources.
Specialized Database Characteristics and Applications
Different research objectives require specialized database features. For research focusing specifically on human protein interactions, HPRD provides not only information on protein interactions but also a variety of protein-specific information, such as post-translational modifications, disease associations, and enzyme-substrate relationships [24]. This integrated approach makes it particularly valuable for clinical and translational research contexts.
For studies requiring the most current data from high-throughput studies, IntAct represents one of the most comprehensive options, though it cites fewer publications (approximately 3,000) compared to HPRD (over 18,000 publications) or BioGRID (16,369 publications) [24]. This suggests different curation priorities, with IntAct concentrating on high-throughput studies while HPRD incorporates more small-scale publications.
The coverage of certain databases may be skewed for some gene types, and analysis with gold-standard PPI-sets has revealed that GPS-Prot, STRING, APID, and HIPPIE each cover approximately 70% of curated interactions [16]. Importantly, database usage frequencies among researchers do not always correlate with their respective advantages, reinforcing the need for evidence-based selection approaches rather than following convention [16].
Methodological Framework for Database Selection
Experimental Protocol for Database Evaluation
A rigorous approach to database selection begins with defining explicit evaluation criteria aligned with research goals. The following protocol provides a systematic methodology for assessing database suitability:
Phase 1: Requirements Analysis
- Define primary research objectives (discovery vs. validation)
- Identify target organisms and specific protein families of interest
- Determine required evidence levels (experimental only vs. including predictions)
- Establish necessary metadata requirements (experimental methods, confidence scores, etc.)
Phase 2: Representative Query Set Development
- Select 100+ genes representative of research focus [16]
- Include well-studied and less-studied genes to assess coverage breadth
- Incorporate genes with different expression patterns (tissue-specific, ubiquitous)
- For human studies, include disease-associated genes relevant to research focus
Phase 3: Comparative Coverage Assessment
- Execute standardized queries across candidate databases
- Record quantitative metrics: total interactions, unique interactions, evidence types
- Assess qualitative factors: annotation depth, data freshness, identifier mapping
- Document practical considerations: API accessibility, download formats, rate limits
Phase 4: Validation Against Gold Standards
- Compare retrieved interactions against literature-curated, experimentally-proven PPI sets [16]
- Calculate precision and recall metrics for each database
- Identify systematic gaps or biases in coverage
Phase 5: Combination Optimization
- Analyze overlap and complementarity between databases
- Identify optimal combinations for specific research goals
- Establish workflow for integration and redundancy management
Database Selection Methodology Workflow
Database Integration and Interoperability Strategies
Effective use of multiple databases requires strategies to address interoperability challenges. Differences in data annotation, protein identifier systems, and interaction evidence representation create significant integration barriers [24]. The International Molecular Exchange (IMEx) consortium aims to enable data exchange and avoid duplication of curation effort through the proteomics standards initiative - molecular interaction (PSI-MI) standard [24]. However, researchers often still need to combine PPI data from all available databases using custom scripts to obtain comprehensive networks [24].
Table 2: Database Combination Strategies for Common Research Scenarios
| Research Goal | Recommended Primary Databases | Supplemental Databases | Rationale |
|---|---|---|---|
| Comprehensive human PPI discovery | STRING, UniHI [16] | HPRD, IID | Covers ~84% of experimentally verified interactions [16] |
| Maximum total interaction coverage | hPRINT, STRING, IID [16] | BioGRID, IntAct | Retrieves ~94% of total available PPIs [16] |
| High-confidence experimental interactions | GPS-Prot, STRING, APID, HIPPIE [16] | MINT, DIP | Each covers ~70% of curated interactions [16] |
| Literature-focused curation | HPRD, BioGRID [24] | IntAct, MINT | Extensive publication coverage (HPRD: 18,777; BioGRID: 16,369) [24] |
When integrating data from multiple sources, particular attention must be paid to identifier mapping consistency. Studies have found that even for the same publications, different databases may report different numbers of interactions [24]. For example, for one publication reporting extensive interactions, different databases listed between 18,877 and 20,800 interactions, with variations potentially attributable to identifier mapping problems [24]. These discrepancies highlight the importance of transparent data processing pipelines and careful handling of protein identifiers.
Research Reagent Solutions for PPI Database Analysis
Table 3: Essential Computational Tools for PPI Database Research
| Tool Category | Specific Solutions | Primary Function | Application Context |
|---|---|---|---|
| Database Access APIs | BioGRID API, STRING API, IntAct Web Services | Programmatic data retrieval | Automated workflow integration; large-scale data extraction |
| Identifier Mapping | UniProt ID Mapping, BioDBnet | Cross-referencing protein identifiers | Solving interoperability challenges between databases |
| Interaction Visualization | Cytoscape, NetworkX | Network visualization and analysis | Exploratory data analysis; result interpretation and presentation |
| Data Integration Platforms | APID (Agile Protein Interaction DataAnalyzer), ConsensusPathDB | Meta-database access | Access to pre-integrated interaction datasets from multiple sources |
| Standardized Formats | PSI-MI XML, MITAB | Standardized data exchange | Facilitating consistent data interpretation and software interoperability |
Advanced Technical Considerations
Experimental Method Biases in PPI Data
Different experimental techniques produce fundamentally different interaction data, and database coverage varies significantly across method types. Yeast two-hybrid (Y2H) systems assay whether two proteins physically interact with each other using genetically modified yeast strains [24]. In contrast, affinity purification followed by mass spectrometry (AP-MS) identifies whole protein complexes rather than pairwise interactions [24]. The representation of AP-MS data can follow either the matrix model (assuming all proteins in a purified complex interact) or the spokes model (assuming interactions only between the tagged protein and each co-purified protein) [24]. These methodological differences significantly impact network topology and must be considered when selecting databases for specific research applications.
PPI Experimental Method Data Representations
Temporal Dynamics and Data Freshness
PPI databases exhibit significant variation in update frequency and data currency. Research comparing six major databases found substantial differences in how quickly new interactions are incorporated and how comprehensively literature is curated [24]. Some databases focus on rapid inclusion of high-throughput datasets, while others prioritize depth of curation from diverse publication types. The synchronization of data between resources participating in the IMEx consortium has improved but remains incomplete, necessitating continued multi-source searching for comprehensive coverage [24].
Optimal database selection in PPI research requires a nuanced approach that aligns technical capabilities with specific research objectives. Evidence demonstrates that strategic database combinations outperform single-source approaches, with specific pairings achieving 84-94% coverage of available interaction data [16]. Researchers should implement the systematic evaluation methodology outlined in this guide, selecting database combinations based on quantitative coverage assessments rather than convention or convenience alone.
Future developments in data standardization through the IMEx consortium and PSI-MI standards promise to reduce current interoperability challenges [24]. However, the evolving nature of protein interaction research ensures that database selection will remain a critical, ongoing consideration in research design. By establishing systematic evaluation protocols and maintaining awareness of the specialized strengths of different resources, researchers can maximize the yield and reliability of their PPI investigations while minimizing biases introduced by incomplete or non-representative data sourcing.
Conclusion
Effective utilization of PPI data requires a nuanced approach that strategically combines multiple databases, applies rigorous validation, and tailors networks to specific biological contexts. The k-votes integration method and the construction of tissue-specific networks represent significant advancements over using single databases or simple unions, leading to more reliable and biologically insightful results. As the field evolves, future directions will be shaped by the increased integration of AI and machine learning for automated data extraction and validation, the growth of more sophisticated tissue- and condition-specific network models, and a stronger emphasis on standardized benchmarking. These developments will further solidify the role of PPI networks as an indispensable tool for unraveling disease mechanisms and accelerating therapeutic development.
References
- 1. About the BioGRID [http://wiki.thebiogrid.org/doku.php/aboutus]
- 2. The BioGRID database: A comprehensive biomedical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7737760/]
- 3. HPRD - Database Commons [https://ngdc.cncb.ac.cn/databasecommons/database/id/1383]
- 4. HPRD -- Human Protein Reference Database | HSLS [https://www.hsls.pitt.edu/obrc/index.php?page=URL1055173331]
- 5. The Molecular INTeraction Database – An ELIXIR Core ... [https://mint.bio.uniroma2.it/]
- 6. DIP: the Database of Interacting Proteins - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC102387/]
- 7. Item - Human PPI from IntAct database (IAH) - figshare - Figshare [https://figshare.com/articles/dataset/Human_PPI_from_IntAct_database_IAH_/5674858]
- 8. Home - Reactome Pathway Database [https://reactome.org/]
- 9. Reactome: a database of reactions, pathways and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3013646/]
- 10. BioGRID Curation Guide [http://wiki.thebiogrid.org/doku.php/curation_guide]
- 11. The BioGRID interaction database: 2019 update - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6324058/]
- 12. BioGRID Curation Workflow [http://wiki.thebiogrid.org/doku.php/curation_description]
- 13. Curation Workflow | BioGRID - Help and Support Resources [http://wiki.thebiogrid.org/doku.php/orcs:curation_description]
- 14. The IntAct molecular interaction database in 2012 - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3245075/]
- 15. APID database: redefining protein–protein interaction ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6354026/]
- 16. Systematic comparison of the protein-protein interaction ... [https://www.sciencedirect.com/science/article/pii/S1532046420300071]
- 17. Protein-protein interaction databases - OpenWetWare [https://openwetware.org/wiki/Protein-protein_interaction_databases]
- 18. InterMitoBase: An annotated database and analysis platform of... [https://bmcgenomics.biomedcentral.com/articles/10.1186/1471-2164-12-335]
- 19. The BioC-BioGRID corpus: full text articles annotated for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5225395/]
- 20. Experimental Evidence Codes | BioGRID [http://wiki.thebiogrid.org/doku.php/experimental_systems]
- 21. Build Statistics (4.4.241) - January 2025 | BioGRID [http://wiki.thebiogrid.org/doku.php/build_4.4.241]
- 22. BioGRID | Database of Protein, Chemical, and Genetic ... [http://thebiogrid.org/]
- 23. The STRING database in 2025: protein networks with ... [https://pubmed.ncbi.nlm.nih.gov/39558183/]
- 24. Protein-protein interaction databases: keeping up with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3500230/]
- 25. - Wikipedia Human Protein Reference Database [https://en.wikipedia.org/wiki/Human_Protein_Reference_Database]
- 26. Role for Protein - Protein Interaction Databases in Human Genetics... [https://www.medscape.com/viewarticle/715684_5]
- 27. Reconstruction of the experimentally supported human ... protein [https://pmc.ncbi.nlm.nih.gov/articles/PMC4015887/]
- 28. Recent advances in deep learning for protein-protein interaction [https://pmc.ncbi.nlm.nih.gov/articles/PMC12168265/]
- 29. Welcome to the BioGRID Download File Repository [https://downloads.thebiogrid.org/BioGRID/Latest-Release/]
- 30. BioGRID Downloads: BioGRID Download File Repository [https://downloads.thebiogrid.org/]
- 31. IntAct - Database Commons [https://ngdc.cncb.ac.cn/databasecommons/database/id/534]
- 32. BioGRID Download Format Details [http://wiki.thebiogrid.org/doku.php/downloads]
- 33. Visualization of protein interaction networks: problems and ... [https://pubmed.ncbi.nlm.nih.gov/23368786/]
- 34. Visualization of protein interaction networks: problems and ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-14-S1-S1]
- 35. The STRING database in 2025: protein networks with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11701646/]
- 36. Cytoscape: An Open Source Platform for Complex Network ... [https://cytoscape.org/]
- 37. Integrated network analysis platform for protein- ... [https://www.lifescience.net/publications/1552628/integrated-network-analysis-platform-for-protein-p/]
- 38. Constructing a robust protein-protein interaction network by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3236850/]
- 39. Protein Interaction Analysis [http://www.geneinfinity.org/sp/sp_proteininteraction.html]
- 40. Voting-based integration algorithm improves causal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7869988/]
- 41. Iterative Learning for K-Approval Votes in Crowdsourcing ... [https://www.mdpi.com/2076-3417/11/2/630]
- 42. SPECTRA: An Integrated Knowledge Base for Comparing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4424906/]
- 43. The TissueNet v.2 database: A quantitative view of protein- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5210565/]
- 44. BioGRID Version 5.0.251 Released [https://thebiogrid.org/news/335]
- 45. A tissue-specific atlas of protein–protein associations ... [https://www.nature.com/articles/s41587-025-02659-z]
- 46. https://www.frontiersin.org/journals/bioengineering-and- ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2015.00058/xml/nlm?isPublishedV2=False]
- 47. Construction and Analysis of Protein-Protein Interaction ... [https://www.nature.com/articles/s41598-019-41552-z]
- 48. Construction and analysis of protein-protein interaction ... [https://www.sciencedirect.com/science/article/abs/pii/S0010482524012411]
- 49. The Atlas of Protein–Protein Interactions in Cancer (APPIC) [https://pmc.ncbi.nlm.nih.gov/articles/PMC11734624/]
- 50. Construction and analysis of protein–protein interaction ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2834675/]
- 51. Sequence-Based Protein–Protein Interaction Prediction and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12468386/]
- 52. How to build and analyze biological networks? [https://bioinfo.cd-genomics.com/how-to-build-and-analyze-biological-networks.html]
- 53. A structural approach for finding functional modules from large ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2537570/]
- 54. STRING: functional protein association networks [https://string-db.org/]
- 55. Mining functional gene modules by multi-view NMF of ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-024-11120-5]
- 56. probing disease mechanisms using model systems - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3706760/]
- 57. New insights into protein–protein interaction modulators in ... [https://www.nature.com/articles/s41392-024-02036-3]
- 58. Identifying disease genes by integrating multiple data sources [https://bmcmedgenomics.biomedcentral.com/articles/10.1186/1755-8794-7-S2-S2]
- 59. Introducing dysfunctional Protein-Protein Interactome (dfPPI) [https://www.sciencedirect.com/science/article/pii/S0959440X24001131]
- 60. Drug Target Protein-Protein Interaction Networks [https://pmc.ncbi.nlm.nih.gov/articles/PMC5485489/]
- 61. Context-specific interactions in literature-curated protein ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-018-5139-2]
- 62. Sensitivity analysis on protein-protein interaction networks ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-025-06140-1]
- 63. Heterogeneous network approaches to protein pathway ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11260399/]
- 64. Mitigating false negatives in imbalanced datasets [https://www.sciencedirect.com/science/article/abs/pii/S0957417424025417]
- 65. ChemFH: an integrated tool for screening frequent false ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11223804/]
- 66. Recent Advances and Application of Machine Learning for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12641681/]
- 67. Rapid LC-MS Based High-Throughput Screening Method ... [https://www.nature.com/articles/s41598-017-08602-w]
- 68. Bias recognition and mitigation strategies in artificial ... [https://www.nature.com/articles/s41746-025-01503-7]
- 69. Systematic literature review on bias mitigation in ... [https://link.springer.com/article/10.1007/s43681-025-00721-9]
- 70. AtPIN: Arabidopsis thaliana protein interaction network. [https://thebiogrid.org/98194/publication/atpin-arabidopsis-thaliana-protein-interaction-network.html]
- 71. Overview of Protein–Protein Interaction Analysis [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/overview-protein-protein-interaction-analysis.html]
- 72. An Overview of Current Methods to Confirm Protein-Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6204658/]
- 73. How to Analyze Protein-Protein Interaction: Top Lab ... [https://www.creative-proteomics.com/resource/how-study-protein-protein-interaction.htm]
- 74. Predictomes, a classifier-curated database of AlphaFold- ... [https://www.sciencedirect.com/science/article/pii/S1097276525001054]
- 75. Prediction of protein–protein interaction based ... - BMC Biology [https://bmcbiol.biomedcentral.com/articles/10.1186/s12915-025-02359-9]
- 76. A Comprehensive Review of Transformer-based language ... [https://arxiv.org/html/2507.13646v1]
- 77. Recent Advances and Application of Machine Learning for ... [https://pubmed.ncbi.nlm.nih.gov/41283644/?utm_source=FeedFetcher&utm_medium=rss&utm_campaign=None&utm_content=14_xQ7JEOWXKupr5M7dtPXHY7ZktNkYxcSPIiG_LVlE7Ve19ke&fc=None&ff=20251124200135&v=2.18.0.post22+67771e2]
- 78. Advances in Protein-Protein Interaction Prediction: A Deep ... [https://www.frontiersin.org/journals/bioinformatics/articles/10.3389/fbinf.2025.1710937/full]
- 79. Applying modularity analysis of PPI networks to sequenced ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3485987/]
- 80. Modularity in Biological Networks [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2021.701331/full]
- 81. Modularity in Biological Networks - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC8477004/]
- 82. Protein-protein interaction (PPI) network analysis reveals ... [https://www.sciencedirect.com/science/article/pii/S1687157X23010533]
- 83. Network representation and analysis tools [https://www.ebi.ac.uk/training/online/courses/network-analysis-of-protein-interaction-data-an-introduction/building-and-analysing-ppins/network-representation-and-analysis-tools/]
- 84. konnect2prot 2.0: Integrating advanced analytical tools for ... [https://www.sciencedirect.com/science/article/pii/S2001037025002697]
- 85. An experimentally derived confidence score for binary protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2976677/]
- 86. Semantically assessing the reliability of protein interactions [https://www.sciencedirect.com/science/article/abs/pii/S0025556413001739]
- 87. Inferring high-confidence human protein-protein interactions [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-13-79]
- 88. Info - STRING functional protein association networks [https://string-db.org/cgi/info]
- 89. Discovering reliable protein interactions from high ... [https://pubmed.ncbi.nlm.nih.gov/16055319/]
- 90. Measuring rank robustness in scored protein interaction ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-3036-6]
- 91. an intuitive resource for integrating protein-protein interactions [https://biosignaling.biomedcentral.com/articles/10.1186/s12964-020-00554-5]
- 92. RAGPPI: RAG Benchmark for Protein-Protein Interactions ... [https://arxiv.org/abs/2505.23823]
- 93. PLM-interact: extending protein language models to predict ... [https://www.nature.com/articles/s41467-025-64512-w]