Overcoming the Membrane Barrier: Advanced Strategies for Detecting Challenging Protein Interactions
This article provides a comprehensive analysis of the technical limitations and recent advancements in detecting membrane protein interactions, a critical frontier in cell biology and drug discovery.
Overcoming the Membrane Barrier: Advanced Strategies for Detecting Challenging Protein Interactions
Abstract
This article provides a comprehensive analysis of the technical limitations and recent advancements in detecting membrane protein interactions, a critical frontier in cell biology and drug discovery. Aimed at researchers and drug development professionals, it explores the unique challenges posed by transient, low-affinity interactions and the lipid membrane environment. The scope ranges from foundational concepts and a comparative evaluation of classical versus novel methods—including FRET, cryo-EM, and computational predictions—to practical troubleshooting guides and robust validation frameworks. By synthesizing insights from current literature, this review serves as a strategic guide for selecting, optimizing, and validating techniques to achieve reliable and physiologically relevant data on membrane protein interactomes.
The Membrane Interactome Challenge: Why Standard Methods Fall Short
Troubleshooting Guide: Common Experimental Issues
This guide addresses frequent challenges in detecting membrane protein interactions, from stable complexes to transient encounters.
Problem: Low or No Signal in Co-Immunoprecipitation (Co-IP)
| Possible Cause | Discussion & Analysis | Recommendation |
|---|---|---|
| Stringent Lysis Conditions [1] | Strong ionic detergents (e.g., sodium deoxycholate in RIPA buffer) can denature proteins and disrupt protein-protein interactions. | Use a milder lysis buffer (e.g., Cell Lysis Buffer #9803). Always include sonication to ensure nuclear rupture and optimal protein recovery. [1] |
| Low Protein/Modification Expression [1] | The target protein or its post-translational modification (e.g., phosphorylation) may be expressed at low basal levels, falling below the detection limit. | Consult expression databases (e.g., BioGPS, Human Protein Atlas) and literature. Use chemical modulators to enhance expression. Include phosphatase/protease inhibitors in lysis buffer. [1] |
| Epitope Masking [1] | The antibody's binding site on the target protein may be obscured by the protein's native conformation or bound interaction partners. | Use an antibody that recognizes a different epitope on the target protein. Check the product page for epitope information. [1] |
Problem: Multiple Bands or Non-Specific Binding in Western Blot
| Possible Cause | Discussion & Analysis | Recommendation |
|---|---|---|
| Protein Isoforms or PTMs [1] [2] | The target protein may have multiple isoforms, splice variants, or post-translational modifications (e.g., glycosylation, phosphorylation) that alter its molecular weight. | Check antibody specifications for known isoform reactivity. Consult databases like UniProt or PhosphoSitePlus. Use a positive control to identify expected bands. [1] |
| Non-Specific Antibody Binding [1] [2] | Off-target proteins may bind non-specifically to the beads or the IgG of the immunoprecipitating antibody. | Include a bead-only control and an isotype control. If background is high, pre-clear the lysate by incubating with beads alone before the IP. [1] |
| Incomplete Sample Reduction [2] | Incomplete reduction of disulfide bonds can leave high-order protein species that run at unexpected sizes or appear as smears. | Use fresh reducing agents (BME or DTT) in the sample loading buffer and boil samples in SDS for 5-10 minutes. [2] |
Problem: High Background in Western Blot
| Possible Cause | Discussion & Analysis | Recommendation |
|---|---|---|
| Ineffective Blocking [2] | The blocking reagent may be insufficient or incompatible with the antibodies used, leading to non-specific binding across the membrane. | Use 5% non-fat milk or 3% BSA. Avoid milk or BSA when using antibodies derived from goat or sheep. Ensure diluent contains a detergent like Tween-20. [2] |
| Antibody Over-concentration [2] | Using too high a concentration of primary or secondary antibody can saturate the specific signal and increase non-specific binding. | Titrate antibody concentrations to find the optimal signal-to-noise ratio. Dilute the conjugate further to reduce background. [2] |
| Insufficient Washing [2] | Unbound antibodies not removed during washing steps will contribute to a high, uniform background. | Ensure the washing protocol is sufficient in volume, duration, and number of buffer changes. Wash buffers should contain a detergent like Tween-20 (0.05%). [2] |
Problem: Obscured Target Signal at 25 kDa or 50 kDa after IP
| Possible Cause | Discussion & Analysis | Recommendation |
|---|---|---|
| Heavy/Light Chain Interference [1] [2] | The denatured heavy (~50 kDa) and light (~25 kDa) chains of the IP antibody are detected by the secondary antibody in the Western blot, masking targets of similar size. | Use an anti-IgG, Light Chain Specific secondary antibody to avoid detecting the heavy chain. Alternatively, use antibodies from different species for IP and Western blot. [1] [2] |
Frequently Asked Questions (FAQs)
Q1: My Co-IP shows a strong signal in the input control, but I fail to pull down my target protein. What is the most likely cause? A1: The most probable cause is that your lysis conditions are too stringent and are disrupting the protein-protein interaction. Switch from a strong denaturing buffer like RIPA to a milder cell lysis buffer. Also, verify that the antibody for immunoprecipitation is suitable for native IP and that the protein's epitope is accessible [1].
Q2: I see an unexpected band at a much lower molecular weight than my target protein. What should I do? A2: This often indicates protein degradation. Add a fresh, broad-spectrum protease inhibitor cocktail to your lysis buffer during sample preparation. Running a positive control can help confirm whether the band is a specific degradation product or non-specific binding [2].
Q3: My Western blot after IP has a very high background. How can I troubleshoot this? A3: First, ensure your blocking step was effective and that you are using an appropriate blocking agent. Second, titrate your primary and secondary antibody concentrations, as over-concentration is a common cause of background. Finally, increase the number and volume of washes with a buffer containing Tween-20 [2].
Q4: How can I confirm that a negative result in my interaction assay is genuine and not a technical failure? A4: A comprehensive set of controls is essential. Always include:
- A positive input lysate control to confirm protein expression and antibody functionality.
- A bead-only control to identify non-specific binding to the beads.
- An isotype control to reveal non-specific binding to the IgG of the IP antibody [1].
Experimental Protocol: Co-Immunoprecipitation for Membrane Protein Complexes
This protocol is designed to preserve labile interactions often found in membrane protein complexes.
1. Cell Lysis and Extraction
- Harvest and wash cells with cold PBS.
- Lyse cells using a non-denaturing, mild lysis buffer (e.g., Cell Lysis Buffer #9803). Do not use RIPA buffer for interaction studies. [1]
- Add inhibitors: Supplement the lysis buffer with protease and phosphatase inhibitors immediately before use (e.g., 2.5 mM sodium orthovanadate for tyrosine phosphatases, or a commercial inhibitor cocktail) [1].
- Incubate on ice for 10-30 minutes.
- Sonicate the lysate briefly on ice to shear DNA, reduce viscosity, and ensure complete nuclear and membrane rupture. This step is crucial for maximum protein recovery [1].
- Clarify the lysate by centrifugation at >12,000 x g for 10 minutes at 4°C. Transfer the supernatant to a new tube.
2. Pre-clearing (Optional but Recommended)
- Incubate the clarified lysate with the bare beads (e.g., Protein A/G) for 30-60 minutes at 4°C with gentle agitation.
- Centrifuge to pellet the beads and transfer the pre-cleared supernatant to a new tube. This step reduces non-specific background [1].
3. Immunoprecipitation
- Add the recommended amount of specific antibody or isotype control antibody to the pre-cleared lysate.
- Incubate for 1-2 hours at 4°C with gentle rotation.
- Add the appropriate bead slurry (e.g., Protein A or G beads. Note: Protein A has higher affinity for rabbit IgG; Protein G for mouse IgG) [1].
- Incubate for an additional 1-2 hours (or overnight) at 4°C with rotation.
4. Washing and Elution
- Pellet the beads by brief centrifugation and carefully remove the supernatant.
- Wash the bead-antibody-protein complex 3-4 times with 1 mL of cold lysis buffer (or a mild wash buffer), resuspending completely each time.
- After the final wash, completely remove the wash buffer.
- Elute the bound proteins by adding 2X Laemmli sample buffer and boiling for 5-10 minutes. The sample is now ready for Western blot analysis.
The Scientist's Toolkit: Research Reagent Solutions
| Reagent / Material | Function & Application |
|---|---|
| Mild Cell Lysis Buffer (e.g., #9803) | Extracts proteins under non-denaturing conditions to preserve protein-protein interactions for Co-IP studies. [1] |
| Protease/Phosphatase Inhibitor Cocktails | Prevents the degradation and dephosphorylation of proteins and their modifications during the lysis and IP process, preserving the native state of the interaction. [1] |
| Protein A/G Beads | The solid-phase matrix for immobilizing antibody-antigen complexes. Choice between Protein A (optimal for rabbit IgG) and Protein G (optimal for mouse IgG) can impact binding efficiency. [1] |
| Light Chain Specific Secondary Antibody | Used in Western blot detection after IP to bind only the light chain (~25 kDa) of the primary antibody, preventing the heavy chain (~50 kDa) from obscuring the target protein. [1] [2] |
| ECL Substrate | A chemiluminescent reagent that produces light upon reaction with the HRP enzyme conjugated to the secondary antibody, enabling the detection of the target protein on the blot. [2] |
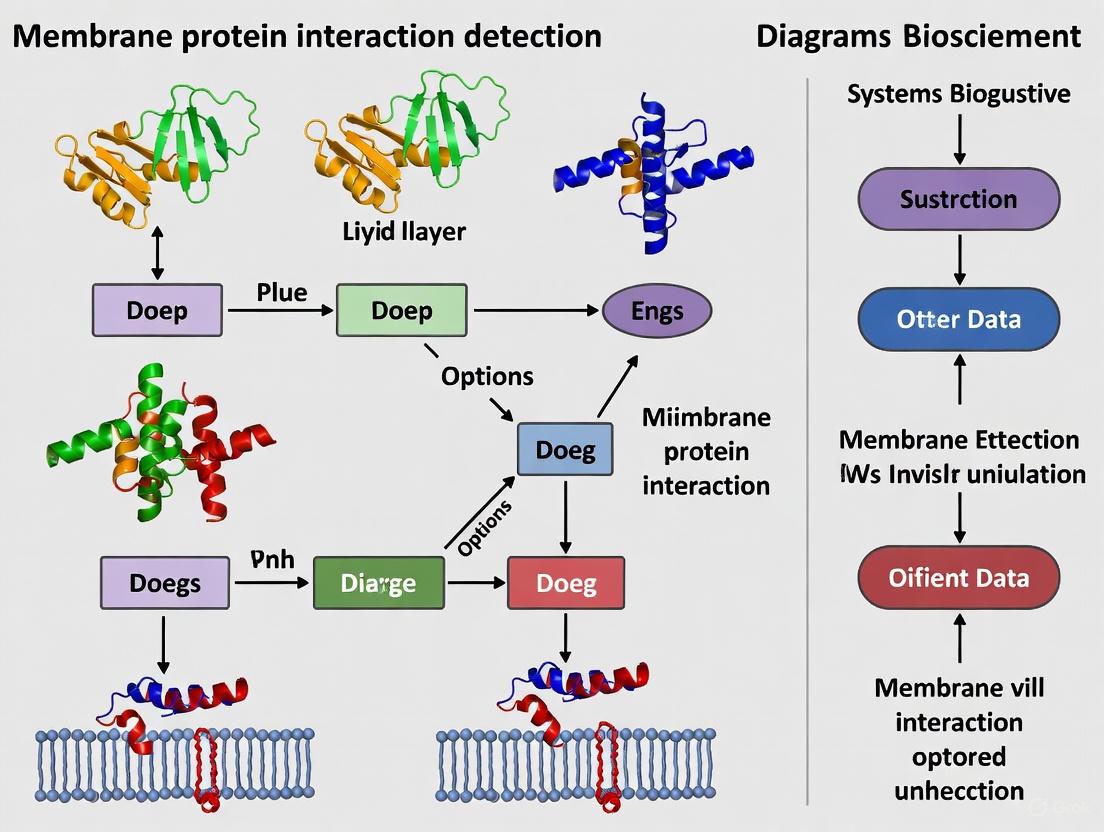
Experimental Workflow and Signaling Diagrams
Core Concepts and Mechanisms
What are the primary mechanisms by which the lipid milieu regulates membrane protein function?
The lipid membrane is an active regulator of protein function, not just a passive barrier. It exerts control through several key mechanisms [3]:
- Direct Interactions (Ligand-like binding): Specific lipids bind to defined sites on proteins, acting as cofactors to stabilize active or inactive conformations.
- Indirect Effects (Biophysical properties): The collective properties of the membrane—such as its fluidity, thickness, elasticity, and lateral pressure—can influence the energy required for proteins to undergo conformational changes during activation.
- Lateral Organization: Membranes segregate into dynamic nano- and microdomains, like cholesterol-enriched lipid rafts and ceramide platforms. Partitioning of proteins into these domains can concentrate partners and modulate signaling activity.
How does 'preferential lipid solvation' differ from specific lipid binding?
A key conceptual advance is the understanding that lipid regulation is not always dependent on long-lived, specific binding sites [4] [5].
- Specific Lipid Binding involves a well-defined, high-affinity interaction between a single lipid molecule and a specific pocket on a protein, often visible in high-resolution structures.
- Preferential Lipid Solvation is a more dynamic and probabilistic process. Certain lipid species are more likely to be found at the protein-lipid interface based on their chemical and physical properties. This creates a local lipid environment that thermodynamically stabilizes certain protein conformations (e.g., associated vs. dissociated dimers) without requiring stable, stoichiometric binding [5].
Frequently Asked Questions (FAQs)
My membrane protein is unstable during extraction and purification. What are my primary options for solubilization?
The choice of solubilization agent is critical and involves a trade-off between stability and experimental applicability. The main options are [6] [7]:
- Detergents: These are the most common choice. They form small micelles around the protein's hydrophobic regions, which is excellent for techniques like X-ray crystallography that require sample homogeneity. However, the detergent environment is chemically distinct from a native membrane and can disrupt protein function and oligomerization [6].
- Nanodiscs / Lipid Polymers: These tools engulf entire sections of the cell membrane, preserving the protein with its native lipid surroundings. This makes them ideal for functional assays as they are more likely to maintain the native oligomerization state and activity. The larger size of the resulting complex may not be suitable for all experimental techniques [6] [7].
My protein expresses poorly or is toxic to my expression system. What can I do?
Low yield and toxicity are common challenges when working with membrane proteins. Consider these troubleshooting steps [6]:
- Change Your Competent Cells: Avoid standard BL21(DE3) E. coli. Use strains like C41(DE3) or C43(DE3), which have mutations that reduce the rate of transcription, making expression less toxic to the host.
- Use a Minimal Growth Medium: Counterintuitively, using a minimal medium like M9, instead of a rich broth, can slow the cell growth rate. This reduces the burden on the protein-folding machinery and can decrease peptide misfolding errors in the membrane.
- Express a Homolog: If your protein of interest is persistently problematic, try expressing a homologous gene from another species. Subtle differences in the primary sequence can confer significant improvements in stability and expression yield.
I am struggling with low binding efficiency during affinity purification. How can I improve this?
When using nickel-affinity or similar chromatography, the solubilizing agent can interfere. Here are some solutions [6]:
- Use a Loose Resin: Perform batch binding by physically mixing a loose resin with your sample for several hours. This allows better access to the affinity tag than a static column.
- Dilute Your Sample: Diluting the sample 2-fold or more can reduce the concentration of the solubilizing agent (detergent), making the affinity tag more accessible for binding.
- Adjust the Affinity Tag: If the tag is buried, consider moving it to the opposite terminus of the protein or lengthening it (e.g., from 6xHis to 12xHis).
Troubleshooting Guides
Problem: Low Functional Yield After Purification
The protein is purified but shows low or no activity in functional assays.
| Possible Cause | Diagnostic Approach | Solution |
|---|---|---|
| Non-native lipid environment (using harsh detergents) | Check literature for known activating lipids. Re-constitute into nanodiscs or liposomes with native lipids and re-test activity [6] [7]. | Switch to a milder detergent for extraction or use a native membrane mimetic like nanodiscs or styrene-maleic acid lipid particles (SMALPs) [6] [8]. |
| Loss of essential lipid cofactor | Use a complementary technique like FIDA to check for ligand binding in different solubilization conditions [7]. | Pre-screen detergents for functional preservation. Add back specific lipids (e.g., cholesterol, phosphoinositides) during or after purification to reconstitute function [3] [4]. |
| Protein denaturation during extraction | Compare the UV chromatogram or dynamic light scattering (DLS) profile before and after extraction for signs of aggregation. | Optimize extraction time and temperature. Sometimes, extraction is more efficient at 20–30°C than at 4°C due to increased thermal motion, but this must be validated for your protein [6]. |
Problem: Inconsistent Results in Ligand-Binding Assays
Binding data is variable or does not match expected physiological behavior.
| Possible Cause | Diagnostic Approach | Solution |
|---|---|---|
| Disruption of lipid-dependent allosteric regulation | Test if the binding affinity of a known ligand is altered in different lipid environments (e.g., cholesterol-depleted membranes) [3] [5]. | Perform binding studies in a more native environment. Flow-Induced Dispersion Analysis (FIDA) allows direct measurement of ligand binding to proteins solubilized in detergents or embedded in nanodiscs, without purification or immobilization [7]. |
| Detergent-induced alteration of the binding site | If possible, compare binding kinetics in detergents vs. nanodiscs. | Use lipid-based membrane mimetics (nanodiscs, liposomes) for binding assays to maintain a native-like conformation of the binding pocket [6] [7]. |
| Ligand accessibility issues in membrane mimetics | Use controls to ensure the ligand can access its site in the chosen system (e.g., in nanodiscs). | Ensure the nanodisc size is appropriate to accommodate the protein and allow ligand access. Characterize the binding in solution to avoid artifacts from surface immobilization [7]. |
Experimental Protocols
Protocol 1: Rapid Detergent Screening for Membrane Protein Stability
Purpose: To quickly identify the optimal detergent for solubilizing and stabilizing a membrane protein using minimal sample [7].
Materials:
- Crude membrane fraction containing your target protein.
- Panel of 12 or more detergents (e.g., DDM, LMNG, OG, Fos-Choline-12).
- Equipment for FIDA or Dynamic Light Scattering (DLS).
Method:
- Prepare Detergent Solutions: Create a matrix of detergent solutions at a concentration above their critical micelle concentration (CMC).
- Solubilize: Add 1.5 µL of your membrane fraction to each detergent condition.
- Incubate: Allow solubilization to proceed for 3 hours at a controlled temperature (e.g., 4°C or 20°C).
- Analyze: Use FIDA or DLS to measure the hydrodynamic radius and polydispersity of the protein in each condition.
- A monodisperse peak indicates a homogeneous, stable sample.
- Aggregation or multiple peaks indicate a poor detergent choice.
- Select: Choose the detergent that yields the smallest hydrodynamic radius and lowest polydispersity, indicating a stable, monodisperse protein population.
Protocol 2: Functional Characterization of a GPCR in Nanodiscs
Purpose: To measure the binding affinity of a ligand to a G-protein coupled receptor (GPCR) while the receptor is embedded in a native-like lipid bilayer (nanodisc) [7].
Materials:
- Purified GPCR reconstituted into nanodiscs.
- Fluorescently labelled or unlabeled ligand of interest.
- FIDA instrument or Surface Plasmon Resonance (SPR) if immobilization is possible.
Method:
- Prepare Sample: Mix the nanodisc-embedded GPCR with varying concentrations of the ligand in solution. No purification or immobilization of the receptor-ligand complex is needed.
- Measure Binding: Use FIDA to monitor the change in the hydrodynamic radius of the nanodisc upon ligand binding. Alternatively, use SPR if a suitable immobilization strategy exists that does not disrupt the nanodisc.
- Quantify Affinity: Plot the change in size (or SPR response) against the ligand concentration.
- Fit Data: Fit the binding isotherm to calculate the dissociation constant (KD), providing a quantitative measure of binding affinity under native-like conditions.
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function / Application | Key Consideration |
|---|---|---|
| C41(DE3) / C43(DE3) E. coli | Expression hosts with reduced transcription rates, ideal for toxic membrane proteins [6]. | Gentler on cells than BL21(DE3), can dramatically improve yield of difficult proteins. |
| n-Dodecyl-β-D-Maltopyranoside (DDM) | Non-ionic detergent, a common first choice for solubilizing membrane proteins while preserving function [6]. | High CMC; forms large micelles. Can be mild but may not preserve all protein-protein interactions. |
| Methyl-β-Cyclodextrin | Cyclic oligosaccharide that efficiently extracts cholesterol from membranes [3]. | Used to study cholesterol-dependent processes; itself has been shown to have analgesic effects via channel inhibition [3]. |
| Nanodiscs (MSP / SAP) | Lipid bilayer discs encircled by a membrane scaffold protein or synthetic polymer, providing a native-like environment [6] [7]. | Excellent for functional studies; size is tunable by the scaffold component. |
| Cobalt-charged Resin | Affinity chromatography resin for purifying His-tagged proteins [6]. | Offers higher purity than nickel-based resins, though with potentially lower sample recovery. |
| Styrene Maleic Acid (SMA) Copolymer | Polymer that directly solubilizes membranes into "SMALPs," preserving a patch of native lipid around the protein [8]. | Allows study of proteins with their native lipid annulus intact. |
Visualization of Concepts and Workflows
Diagram 1: Membrane Protein Regulation Mechanisms
Diagram 2: Membrane Protein Extraction Workflow
Technical Support Center
Troubleshooting Guides
Issue 1: No or Weak Signal in Co-immunoprecipitation (Co-IP) Experiments
- Problem: After performing a co-IP to study protein-protein interactions, the western blot shows little to no signal for the target protein or its interacting partners.
- Potential Causes and Solutions:
- Disrupted Protein Interactions: Lysis buffers that are too stringent (e.g., RIPA buffer containing ionic detergents like sodium deoxycholate) can denature proteins and disrupt weak interactions [9]. Solution: Use a milder lysis buffer (e.g., Cell Lysis Buffer #9803) and include sonication to ensure adequate protein recovery without disrupting complexes [9].
- Low Protein Abundance: The target protein or its post-translationally modified forms (e.g., phosphorylated proteins) may be expressed at very low levels [9]. Solution: Check protein expression profiles using databases. Include an input lysate control (1-10% of your starting lysate) to confirm the target is present and detectable. For phosphorylated proteins, use phosphatase inhibitors in your lysis buffer [9] [10].
- Epitope Masking: The antibody's binding site might be blocked by the protein's conformation or other interacting proteins [9]. Solution: Try an antibody that recognizes a different epitope on the target protein [9].
- Inefficient Antibody-Bead Binding: The wrong bead type can reduce binding efficiency [9]. Solution: Use Protein A beads for rabbit antibodies and Protein G beads for mouse antibodies, or use combination A/G beads [9].
Issue 2: High Background or Non-Specific Bands in Western Blot
- Problem: The western blot membrane has a high background or shows multiple non-specific bands, making results difficult to interpret.
- Potential Causes and Solutions:
- Non-Specific Binding to Beads: Proteins can stick to the beads or the IgG antibody itself [9]. Solution: Include a bead-only control (beads with no antibody) and an isotype control (a non-specific antibody of the same IgG class). Pre-clear the lysate by incubating with beads alone before the IP [9].
- Antibody Cross-Reactivity: The primary antibody may detect multiple isoforms or post-translationally modified versions of the target protein, or unrelated proteins with similar epitopes [9] [2]. Solution: Run an input control to see if the extra bands are present in the original lysate. Consult antibody datasheets and scientific databases to check for known isoform reactivity or modifications [9].
- Ineffective Blocking or Washing: Solution: Ensure the membrane is properly blocked with 5% non-fat milk or 3% BSA. Use sufficient wash volume, time, and number of changes in wash buffer containing 0.05% Tween 20 [2].
- Antibody Overconcentration: Solution: Titrate the primary and secondary antibody concentrations to find the optimal signal-to-noise ratio [2].
Issue 3: IgG Heavy/Light Chains Obscuring the Target Protein Band
- Problem: During western blot analysis after IP, the heavy (~50 kDa) and light (~25 kDa) chains of the IP antibody obscure the detection of your protein of interest if it migrates to a similar molecular weight.
- Potential Causes and Solutions:
- Same Host Species for IP and Blot Antibodies: The secondary antibody detects the denatured IgG chains from the IP antibody [9] [2].
- Solutions:
- Use primary antibodies from different species for the IP and the western blot (e.g., rabbit for IP, mouse for blot, or vice-versa) [9] [2].
- Use a light-chain specific secondary antibody for detection, which will not recognize the heavy chain [9] [2].
- Use a biotinylated primary antibody for blotting and detect with Streptavidin-HRP [9].
Frequently Asked Questions (FAQs)
Q1: What is the critical control I need to include in every Co-IP experiment? A1: The most critical control is the Input Lysate. This is 1-10% of your original cell or tissue lysate, set aside before adding any antibodies or beads. It confirms that your target protein is present and detectable in the starting material, providing a benchmark for the efficiency of your immunoprecipitation [10].
Q2: My membrane protein of interest is difficult to work with. What are some advanced techniques to study its interactions? A2: Traditional methods like co-IP can be challenging for membrane proteins. Advanced techniques that are powerful for this purpose include:
- Native Mass Spectrometry (nMS): Analyzes intact protein complexes under non-denaturing conditions, providing information on molecular mass, oligomeric state, and lipid/protein interactions [11].
- Single-Molecule Fluorescence Methods: Techniques like Fluorescence Correlation Spectroscopy (FCS) and super-resolution microscopy (e.g., STED, PALM) can quantify protein association states and spatial organization in the native membrane environment of live cells [12].
- Mass Photometry: Rapidly measures the mass of individual molecules in solution, providing insights into sample homogeneity, oligomerization, and the effects of different membrane mimetics (detergents, nanodiscs) on protein stability [13].
Q3: What are the main limitations of Co-IP, and how can I address them? A3: Co-IP has several key limitations [10]:
- Transient or Weak Interactions: It may not capture interactions that are brief or of low affinity. Solution: Use crosslinking to stabilize these interactions before lysis.
- Indirect Interactions: It cannot distinguish between proteins that bind directly to your bait protein and those that are part of a larger complex (indirect interactors). Solution: Validate interactions with a complementary technique, such as pull-down assays with purified components.
- Antibody Interference: The antibody used for IP might bind to the interaction site, blocking prey protein binding. Solution: Test antibodies against different epitopes or use a tagged version of your protein.
Q4: How do I choose a lysis buffer for my Co-IP experiment? A4: The choice is critical and depends on the interaction strength [9]:
- For strong, stable complexes, a milder, non-denaturing lysis buffer (e.g., Cell Lysis Buffer #9803) is suitable.
- For weak or transient complexes, avoid strong ionic detergents (like those in RIPA buffer) as they will disrupt the interaction.
- Always include protease and phosphatase inhibitors in your lysis buffer to preserve the protein's state.
Experimental Data and Protocols
Table 1: Common Issues and Quantitative Solutions in Co-IP/Western Blotting
| Problem | Possible Cause | Recommended Solution | Positive Control Check |
|---|---|---|---|
| Low/No Signal | Stringent lysis buffer disrupting interactions [9] | Switch to mild cell lysis buffer; Include sonication [9] | Input lysate [9] [10] |
| Low abundance of target protein [9] | Use >300 µg total protein; enrich via subcellular fractionation [10] | BioGPS / Protein Atlas database check [9] | |
| Low phospho-protein levels [9] | Use phosphatase inhibitors (Na pyrophosphate, β-glycerophosphate, Na orthovanadate) [9] | PhosphoSitePlus check; use known modulators [9] | |
| High Background | Non-specific bead binding [9] | Pre-clear lysate; use bead-only control [9] | Isotype control [9] |
| Antibody cross-reactivity [2] | Titrate antibody concentration; check specificity with negative control lysate [2] | Negative control (non-transfected cell lysate) [2] | |
| IgG Masking | Same species for IP and blot antibodies [9] [2] | Use different species for IP and blot; use light-chain specific secondary [9] [2] | Check target protein molecular weight vs. IgG chains (25/50 kDa) |
Essential Research Reagent Solutions
| Item | Function / Explanation | Example / Key Consideration |
|---|---|---|
| Cell Lysis Buffer #9803 | A mild lysis buffer recommended for Co-IP to preserve native protein-protein interactions, unlike more denaturing RIPA buffer [9]. | Suitable for co-IP experiments [9]. |
| Protein A & G Beads | Solid support for immobilizing antibodies to pull down the antigen. Protein A has higher affinity for rabbit IgG; Protein G for mouse IgG [9]. | Optimize bead choice based on antibody host species [9]. |
| Protease/Phosphatase Inhibitor Cocktail | Added to lysis buffers to prevent protein degradation and maintain post-translational modifications (e.g., phosphorylation) during sample preparation [9]. | Essential for studying labile modifications [9]. |
| Light Chain Specific Secondary Antibody | Used in western blotting after IP to detect the primary antibody without detecting the heavy chain of the IP antibody, preventing masking of targets at ~50 kDa [9] [2]. | Anti-Rabbit IgG (Light-Chain Specific) mAb #93702 [9]. |
| Membrane Mimetics (Nanodiscs, Amphipols) | Systems used to solubilize and stabilize membrane proteins in a near-native lipid environment for biophysical analysis like native MS or cryo-EM [11] [13]. | Crucial for maintaining the structure and function of integral membrane proteins outside the cell membrane [13]. |
| Biotinylated Antibodies | Used in innovative antibody-uptake assays to track endocytosis of membrane proteins. Detected with high-affinity streptavidin-HRP on western blots [14]. | High-affinity bond (KD 10−14-10−16 M) allows robust detection where traditional methods fail [14]. |
Experimental Workflow and Pathway Visualization
Co-Immunoprecipitation (Co-IP) Workflow
Advanced Techniques for Membrane Protein Interaction Studies
FAQs: Addressing Critical Challenges in Membrane Protein Research
Q1: How does my choice of lysis buffer risk disrupting membrane protein interactions during co-immunoprecipitation?
The stringency of your lysis buffer is a critical factor. Strong denaturing buffers can dissociate native protein complexes, leading to false-negative results in co-IP experiments.
- Problem: Using RIPA buffer, which contains ionic detergents like sodium deoxycholate, is suitable for whole-cell extracts for western blotting but is known to denature kinases and prevent protein-protein interactions. This makes it unsuitable for co-IP studies where preserving native complexes is the goal [15].
- Solution: For co-IP experiments, use a milder, non-denaturing lysis buffer. A recommended starting point is a cell lysis buffer that lacks harsh ionic detergents. Ensure proper sonication during lysis to aid in nuclear rupture and membrane protein extraction without disrupting most protein complexes [15].
Q2: My target membrane protein interaction is transient with low binding affinity. What methods can capture these elusive complexes?
Low-affinity interactions (with dissociation constants, K_D, in the micromolar range or with half-lives of less than a second) are common in signaling and immune recognition. Traditional methods often fail because complexes dissociate during wash steps [16] [17]. The table below summarizes advanced strategies to overcome this.
- Avidity-Enhanced Screening: The AVEXIS (Avidity-Based Extracellular Interaction Screen) assay is designed specifically for this purpose. It uses a pentamerized "prey" protein to increase avidity, allowing it to detect extremely transient interactions with half-lives as short as 0.1 seconds [17].
- Molecular Engineering: Techniques like single-chain fusions and disulfide trapping (discussed in detail in the Experimental Protocols section) can covalently stabilize weak complexes for structural studies like X-ray crystallography or cryo-EM [16].
Table 1: Strategies for Capturing Low-Affinity Protein Interactions
| Strategy | Principle | Best Suited For | Key Advantage |
|---|---|---|---|
| AVEXIS [17] | Uses pentamerized prey proteins to multiplicatively enhance binding avidity to immobilized monomeric bait. | Systematic, high-throughput screening of extracellular receptor-ligand pairs. | Can detect interactions with half-lives ≤ 0.1 sec; low false-positive rate. |
| Single-Chain Fusions [16] | Genetically links two protein partners with a flexible peptide linker, enforcing proximity and high local concentration. | Stabilizing complexes for structural biology (e.g., TCR-pMHC, SRP-FtsY). | Simple design; ensures equimolar presence of both partners. |
| Disulfide Trapping [16] | Introduces cysteine residues at binding interfaces to form covalent disulfide bonds upon co-incubation. | Mapping interfaces and stabilizing defined complexes (e.g., GPCR-ligand structures). | Provides covalent stabilization; useful for mapping residue proximity. |
| Chemical Crosslinking [18] | Adds membrane-permeable or impermeable bifunctional reagents to covalently "freeze" interactions inside or outside the cell. | Capturing putative interacting partners in situ before lysis. | Can be applied in live cells; suitable for transient, intracellular interactions. |
Q3: What is the most effective workflow to detect low-abundance proteins masked by high-abundance serum proteins?
In complex samples like serum, high-abundance proteins like albumin and immunoglobulins can obscure the detection of low-abundance biomarkers. A robust, multi-step enrichment workflow is required.
- Immunodepletion of IgG: Use a Protein G column to efficiently remove immunoglobulins (IgG), which constitute over 35% of serum protein content [19].
- Albumin Reduction via Preparative Electrophoresis: Subject the IgG-depleted serum to denaturing preparative gel electrophoresis in the presence of SDS. This strong ionic detergent prevents the "sponge effect" where albumin non-specifically binds other proteins. The albumin band is localized and excised, significantly enriching the low-abundance proteins in the remaining fractions [19].
- Sensitive Downstream Detection: Analyze the enriched sample using sensitive methods like nLC-MS/MS. For western blotting, use high-sensitivity chemiluminescent substrates to detect the now-unmasked low-abundance targets [19] [20].
Workflow for Serum Protein Enrichment
Experimental Protocols
Protocol 1: AVEXIS for Detecting Low-Affinity Extracellular Interactions
Purpose: To systematically identify very transient (half-life ≤ 0.1 sec), low-affinity interactions between extracellular protein domains [17].
Workflow:
AVEXIS Assay Workflow
- Protein Production:
- Express the entire ectodomains of your proteins of interest in a mammalian system to ensure proper post-translational modifications.
- Generate two forms:
- Bait: A monomeric, biotinylated version.
- Prey: A pentamerized version, created by C-terminal fusion to a coiled-coil sequence from rat cartilage oligomeric matrix protein, and tagged with an enzyme like β-lactamase for detection.
- Activity Normalization: Normalize the activity/concentration of all bait and prey proteins before screening.
- Screening Assay:
- Capture the biotinylated bait protein on a streptavidin-coated microtiter plate.
- Incubate with the pentamerized prey protein.
- Wash the plate to remove unbound prey.
- Detect specifically bound prey by measuring the activity of the β-lactamase tag.
- Validation: Re-test all potential hits in the reciprocal orientation (i.e., the original prey protein used as bait and vice versa). Only consider interactions that are positive in both orientations as high-confidence hits [17].
Protocol 2: Enriching Low-Abundance Proteins from Serum via Preparative Electrophoresis
Purpose: To remove high-abundance albumin and immunoglobulins from serum to enable the identification and quantification of low-abundance proteins [19].
- Depletion of Immunoglobulins:
- Dilute serum sample 5-fold in a suitable binding buffer (e.g., 20 mM sodium phosphate, pH 7.0).
- Load the diluted serum onto a Protein G column pre-equilibrated with the same buffer.
- Collect the flow-through as the IgG-depleted serum.
- Denaturing Preparative Gel Electrophoresis:
- Mix the IgG-depleted serum with SDS-PAGE loading dye and heat at 62°C for 20 minutes.
- Load the sample (e.g., 15 mg of protein) onto a denaturing preparative polyacrylamide gel (e.g., a stepwise gel with layers of 15%, 12%, and 10% polyacrylamide).
- Run the gel at a constant current (e.g., 45 mA) until the dye front elutes.
- Continuously elute the separated proteins from the bottom of the gel, collecting small-volume fractions (e.g., 300 fractions of 0.5 mL each).
- Locating and Pooling Albumin-Reduced Fractions:
- Analyze a small aliquot of each fraction by standard SDS-PAGE followed by silver staining.
- Identify fractions that contain proteins but are devoid of the prominent albumin band.
- Pool these albumin-reduced fractions, concentrate, and buffer-exchange them for downstream analysis (e.g., tryptic digestion for nLC-MS/MS) [19].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for Overcoming Technical Hurdles
| Item | Function | Application Context |
|---|---|---|
| Gentle Lysis Buffer (e.g., without ionic detergents like deoxycholate) | Extracts proteins while preserving weak, native protein-protein interactions. | Co-immunoprecipitation (Co-IP) assays [15]. |
| Membrane-Permeable Crosslinker (e.g., DSS - Disuccinimidyl suberate) | Covalently "freezes" transient protein interactions inside live cells before lysis. | Capturing intracellular complexes for pull-downs or Co-IP [18]. |
| Protein G/A/G Beads | High-affinity solid support for binding and precipitating antibody-antigen complexes. | Immunoprecipitation and Co-IP [15]. |
| Protease/Phosphatase Inhibitor Cocktails | Prevents protein degradation and maintains post-translational modifications during sample preparation. | All sample preparation steps, especially for labile proteins and phospho-proteins [15] [21]. |
| High-Sensitivity Chemiluminescent Substrate (e.g., SuperSignal West Femto/Atto) | Provides maximum sensitivity for detecting low-abundance proteins on western blots, down to the attogram level. | Western blot detection of rare targets or from limited sample material [20] [21]. |
| Tris-Acetate Gels | Optimal separation and transfer of high molecular weight proteins (>300 kDa) by allowing better migration into the gel matrix. | Gel electrophoresis of large membrane proteins [20]. |
| Tricine Gels | Provides superior resolution of low molecular weight proteins (<30 kDa). | Gel electrophoresis of small protein subunits or cleavage products [20]. |
A Modern Toolkit: From Classical Biochemistry to Cutting-Edge Biophysics
Troubleshooting Guides
Common Co-IP Experimental Issues and Solutions
| Problem | Possible Causes | Recommended Solutions |
|---|---|---|
| Low/No Signal | Stringent lysis buffer disrupting protein-protein interactions [22] | Use mild lysis buffers (e.g., Cell Lysis Buffer #9803); avoid strong ionic detergents like RIPA for Co-IP [22]. |
| Low expression of target or modified protein [22] | Check expression with input control; use bioinformatics tools (e.g., BioGPS) and include positive controls [22]. | |
| Epitope masking by protein conformation or interacting partners [22] | Use an antibody that recognizes a different epitope on the target protein [22]. | |
| Weak or transient protein interactions [23] | Perform all steps at 4°C; use mild buffers; consider crosslinking prior to Co-IP [23]. | |
| High Background/Non-specific Bands | Non-specific binding of proteins to beads or IgG [22] [23] | Include a bead-only control; pre-clear the lysate; use an isotype control antibody [22] [23]. |
| Antibody concentration too high [23] | Optimize antibody concentration by titration [23]. | |
| Washes not stringent enough [23] | Increase number of washes; optimize wash buffer stringency by adjusting salt/detergent concentration [23]. | |
| Prey Not Detected in Pulldown | Prey protein is insoluble or denatured [24] | Optimize lysis and IP conditions specifically for the prey protein [24]. |
| Antibody blocks bait-prey interaction site [23] | Try an alternative antibody that binds a different epitope [23]. | |
| Interaction requires specific additives or components [23] | Include necessary co-factors (if known) in the lysis or IP buffer [23]. | |
| IgG Heavy/Light Chains Obscuring Target | Target protein migrates near 25 or 50 kDa [22] | Use different species for IP and WB antibodies; use light-chain specific secondary antibodies [22]. |
Common Pull-Down Assay Issues and Solutions
| Problem | Possible Causes | Recommended Solutions |
|---|---|---|
| High Background | Non-specific binding to affinity support [25] | Increase number of elution steps; include a negative control with affinity support alone [26] [25]. |
| Add non-ionic detergent (e.g., 0.1% Tween-20); pre-treat beads with specific pH buffer [25]. | ||
| Blurry or Smeared Bands | Insufficient elution conditions [25] | Increase the concentration of the elution buffer [25]. |
| Low purity of the bait protein [25] | Improve the purification protocol for the tagged bait protein [25]. | |
| False Positives | Non-specific adsorption [25] | Pre-clear lysate with bead-only incubation; use competitor proteins like BSA to block non-specific sites [23] [25]. |
| No Interaction Detected | Protein expressed as inclusion bodies [25] | Denature and refold the protein before the assay [25]. |
| Tagged protein degraded [26] | Include protease inhibitors in the lysis buffer [26]. | |
| Low abundance of bait or prey [26] | Use more lysate for the pulldown; employ a more sensitive detection system [26]. |
Frequently Asked Questions (FAQs)
Experiment Design & Controls
What are the essential controls for a Co-IP experiment? A complete Co-IP experiment requires several controls for meaningful interpretation [24]:
- Input Lysate Control: 1-10% of the lysate reserved before IP. Confirms the presence of both bait and prey proteins and serves as a positive control for western blot detection [22] [10].
- Negative Control (Bead-Only): Beads without antibody. Identifies proteins that bind non-specifically to the bead matrix [22].
- Negative Control (Isotype): An irrelevant antibody of the same isotype. Identifies proteins that bind non-specifically to the IgG [22].
- Positive Control: A sample with a known interaction to confirm the Co-IP protocol is working correctly [22].
How do I know if my lysis buffer is suitable for Co-IP? The lysis buffer must be strong enough to solubilize the target proteins but mild enough to preserve their native interactions [22] [23]. Strong denaturing buffers like RIPA (which contains sodium deoxycholate) can disrupt protein-protein interactions and are not recommended for Co-IP. Start with a mild, non-denaturing lysis buffer and include protease and phosphatase inhibitors to maintain protein integrity and post-translational modifications [22] [26].
Can I use the same antibody for immunoprecipitation and western blotting? Yes, but this can cause a problem where the denatured heavy (~50 kDa) and light (~25 kDa) chains of the IP antibody obscure the detection of your target protein on the western blot if they migrate at a similar size [22]. To avoid this:
- Use primary antibodies from different host species for the IP and the WB [22].
- Use a biotinylated primary antibody for WB and detect with Streptavidin-HRP [22].
- Use a light-chain specific secondary antibody for WB [22].
Technical Execution
Why is my bait protein not binding to the beads?
- Antibody-Bead Immobilization: Ensure the bead type (e.g., Protein A vs. Protein G) is compatible with the host species and isotype of your antibody [22] [23]. Protein A has high affinity for rabbit IgG, while Protein G has high affinity for mouse IgG [22].
- Antibody Quality: Confirm the antibody is suitable for immunoprecipitation. Polyclonal antibodies should be affinity-purified to prevent competition from other immunoglobulins [23].
- Solubility: The bait protein may be insoluble or unfolded. Optimize lysis conditions and confirm solubility in the input sample [24].
I see my bait protein but not the prey protein in the final eluate. What went wrong?
- Interaction Disruption: The lysis or wash conditions may be too harsh, disrupting the bait-prey interaction. Try milder buffers and ensure all steps are performed at 4°C [22] [23].
- Prey Solubility: The prey protein itself may be insoluble or denatured [24].
- Epitope Blocking: The antibody used for IP might be binding to an epitope that is critical for the bait-prey interaction, thus blocking it. Try an antibody against a different epitope [23].
- No Interaction: The proteins may not interact under your experimental conditions.
The prey protein is appearing in my negative controls. What does this mean? If the prey protein is precipitated in the absence of the bait protein (e.g., in a bead-only or isotype control), this indicates non-specific binding [24]. To resolve this:
- Increase Wash Stringency: Increase the number of washes or adjust the salt/detergent concentration in the wash buffer [23].
- Pre-clearing: Pre-clear the lysate by incubating it with beads alone before adding the antibody-bound beads [22].
- Block Beads: Block the beads with a competitor protein like BSA [23].
- Use Low-Binding Tubes: Switch to low-binding plastic consumables to reduce adhesion [24].
Methodology & Analysis
What is the key difference between Co-IP and Pull-Down assays?
- Co-IP uses a specific antibody to precipitate a native "bait" protein and its binding partners directly from a cell or tissue lysate. It confirms interactions in a near-physiological context but cannot distinguish between direct and indirect interactions [10] [25].
- Pull-Down Assays use an immobilized "bait" molecule (e.g., a purified tagged protein, DNA, RNA, or lipid) to capture binding partners from a lysate. It can confirm direct interactions but occurs in an artificial, in vitro environment [25].
How can I study interactions involving membrane proteins or lipids? Traditional methods struggle with hydrophobic membrane components. Advanced techniques are now available:
- Lipid-Immobilized Beads: Beads coated with specific lipids (e.g., sphingomyelin, cholesterol) can be used to screen for lipid-binding membrane proteins from detergent-solubilized mixtures in a pull-down format [27].
- Nanodisc Single-Molecule Pulldown (SiMPull): Nanodiscs provide a stable, flat membrane mimic. When loaded with specific lipids, they can be used in pull-down assays to investigate lipid-protein interactions with single-molecule resolution, allowing measurement of binding kinetics [28].
My Co-IP works but my GST Pull-Down does not. Why? This is a common scenario. Co-IP detects interactions that occur within the complex cellular environment, which may involve additional proteins or post-translational modifications. If the GST Pull-Down, which tests for a direct interaction between two purified proteins, is negative, it suggests that the interaction observed in Co-IP is likely indirect and mediated by a third protein present in the cell lysate [25].
Research Reagent Solutions
| Item | Function | Example / Key Note |
|---|---|---|
| Cell Lysis Buffer | Solubilizes proteins while preserving interactions. | Use mild, non-denaturing buffers (e.g., CST #9803); avoid RIPA for Co-IP [22]. |
| Protease/Phosphatase Inhibitors | Prevents degradation and maintains post-translational modifications. | Essential cocktails (e.g., CST #5872) preserve protein phosphorylation and integrity [22] [26]. |
| Protein A/G Beads | Solid support for antibody immobilization. | Choose based on host species: Protein A for rabbit IgG, Protein G for mouse IgG [22]. |
| Common Epitope Tags | Enable IP when target-specific antibodies are unavailable. | FLAG (DYKDDDDK), c-Myc (EQKLISEEDL), HA (YPYDVPDYA), V5 (GKPIPNPLLGLDST) [10]. |
| Crosslinkers (e.g., DSS, BS3) | "Freeze" transient or weak interactions prior to lysis. | Membrane-permeable (DSS) for intracellular; impermeable (BS3) for cell surface [26]. |
| Lipid-Immobilized Beads | Study lipid-protein interactions for membrane proteins. | Beads coated with specific lipids (e.g., sphingomyelin, cholesterol) to mimic membrane environments [27]. |
| Nanodiscs | Membrane mimics for studying lipid-protein interactions. | Provide a stable, flat phospholipid bilayer for robust pull-down assays [28]. |
Experimental Workflows
Co-IP and Pull-Down Assay Workflow
Co-IP Troubleshooting Logic
The study of membrane protein interactions is crucial for understanding cellular signaling, drug action, and receptor function, yet it presents significant technical challenges. Traditional biochemical methods often lack the spatial and temporal resolution to capture these dynamic events in their native cellular environment [12] [29]. Proximity-based luminescence assays, particularly Bioluminescence Resonance Energy Transfer (BRET) and Split-Luciferase Complementation, have emerged as powerful tools that address these limitations by enabling real-time monitoring of protein-protein interactions in live cells [30] [31]. These technologies leverage bioluminescent enzymes, primarily luciferases, to convert molecular proximity into quantifiable optical signals with high sensitivity and minimal background [30] [32]. This technical support center provides comprehensive guidelines for implementing these advanced methodologies, with particular emphasis on their application for membrane protein research in drug discovery and basic science.
BRET (Bioluminescence Resonance Energy Transfer)
BRET is a biophysical technique that monitors proximity between molecules within live cells based on non-radiative energy transfer [31]. The system consists of a donor luciferase enzyme and an acceptor fluorophore tagged to proteins of interest. Upon substrate addition, the luciferase catalyzes a light-producing reaction. When the acceptor is within close proximity (typically <10 nm), energy transfers from the donor to the acceptor, resulting in fluorescence emission at a characteristic wavelength [32] [31]. This mechanism enables quantitative assessment of protein interactions without requiring external light sources, thereby avoiding issues of autofluorescence and photobleaching common in FRET-based approaches [32].
Split-Luciferase Complementation Assay
Split-luciferase complementation assays are based on dividing a luciferase enzyme into two inactive fragments that are fused to potential interacting proteins [30] [29]. Upon interaction between the target proteins, the luciferase fragments are brought into proximity, facilitating their reassembly into a functional enzyme that produces bioluminescence upon substrate addition [30] [33]. This system benefits from an extremely high signal-to-noise ratio and wide dynamic range, making it particularly suitable for detecting weak or transient interactions [30] [29]. Unlike BRET, which reports on proximity, complementation assays directly indicate interaction events through luciferase activity restoration.
Comparative Analysis of Luciferase Systems
Table 1: Characteristics of Commonly Used Luciferase Systems
| Luciferase | Origin/Source | Size (kDa) | Substrate | Emission λmax (nm) | Key Features | Best Applications |
|---|---|---|---|---|---|---|
| NanoLuc | Engineered from Oplophorus gracilirostris | 19 | Furimazine | 460 [30] | Very bright, small size, high stability [31] | NanoBRET, high-sensitivity detection [31] |
| Firefly (FLuc) | Photinus pyralis | 61 | D-Luciferin | 560 [30] | Glow-type kinetics, requires ATP [30] | Whole-animal imaging, transcriptional reporting |
| Renilla (RLuc) | Renilla reniformis | 36 | Coelenterazine | 480 [30] | Native BRET donor, ATP-independent [32] | BRET1, BRET2 applications |
| Akaluc | Engineered from FLuc | 61 | Akalumine | 650 [30] | Red-shifted emission, deep tissue penetration [30] | In vivo imaging applications |
| GLuc | Gaussia princeps | 20 | Coelenterazine | 473 [30] | Secreted luciferase, small size [30] | Extracellular reporting, secretion assays |
Table 2: Comparison of BRET Methodologies
| BRET Method | Donor Luciferase | Acceptor | Donor Emission (nm) | Acceptor Emission (nm) | Key Features |
|---|---|---|---|---|---|
| BRET 1 | RLuc | eYFP | 480 | 530 [32] | Strong signals, long lifetime [32] |
| BRET 2 | RLuc | GFP | 395 | 510 [32] | Better spectral separation, low light emission [32] |
| eBRET 2 | RLuc8 | GFP | 395 | 510 [32] | 5-fold better signal than BRET 2 [32] |
| BRET 3 | Firefly | DsRed | 565 | 583 [32] | Lower autofluorescence, weak signals [32] |
| NanoBRET | NanoLuc | HaloTag Ligand | 460 | 618 [32] | Excellent separation, very bright [32] [31] |
Troubleshooting Guides: Addressing Common Experimental Challenges
Low Signal-to-Noise Ratio
Problem: Weak BRET or complementation signals make results difficult to interpret.
Solutions:
- Optimize expression levels: High acceptor-to-donor ratios (typically 3:1 to 10:1) improve BRET efficiency [31]. For complementation assays, balance expression of both fusion constructs to maximize productive interactions [34].
- Verify fragment orientation: For membrane proteins, ensure luciferase fragments face the same compartment (e.g., cytoplasmic side) to enable complementation [33].
- Check substrate quality: Use fresh substrates prepared at optimal concentrations. Furimazine for NanoLuc systems provides sustained glow-type kinetics, while coelenterazine derivatives may require timing optimization due to flash-type kinetics [30] [31].
- Confirm protein functionality: Validate that fusion tags do not disrupt protein localization or function through control experiments with established interaction partners [29].
High Background Signal
Problem: Elevated background luminescence in negative controls.
Solutions:
- Reduce spontaneous complementation: For split-luciferase assays, optimize split sites and use fragments with low inherent affinity (e.g., NanoBiT system with LgBiT and SmBiT) [29].
- Minimize non-specific interactions: Include linker sequences (typically 10-15 amino acids) between your protein and reporter fragments to reduce steric hindrance [29].
- Control for overexpression artifacts: Use inducible promoters or stable cell lines with near-endogenous expression levels rather than transient transfection when possible [29].
- Validate specificity: Perform competition experiments with untagged proteins or known inhibitors to confirm signal specificity [34] [35].
Inconsistent Results Between Experiments
Problem: Poor reproducibility across technical and biological replicates.
Solutions:
- Standardize transfection protocols: Use consistent DNA quality, transfection reagents, and timing across experiments [36] [33].
- Control cell confluency: Plate cells at consistent density (e.g., 50,000-100,000 cells/well for 96-well format) and maintain uniform culture conditions [36] [33].
- Implement internal controls: Include positive and negative interaction controls in each experiment to normalize between runs [34].
- Optimize measurement timing: For kinetic studies, establish precise timing for substrate addition and measurement relative to treatments [35].
Lack of Expected Interaction Signal
Problem: No detectable BRET or complementation despite literature evidence of interaction.
Solutions:
- Verify fragment orientation: Test both N- and C-terminal fusions for each protein, as terminal positioning dramatically affects interaction capability [29].
- Check protein expression: Confirm expression of both fusion constructs by Western blot or other methods before luminescence assessment [34].
- Assess membrane localization: For membrane proteins, verify proper trafficking and localization using fluorescence microscopy with tagged versions [12].
- Optimize linkers: Vary linker length and flexibility (e.g., GSG repeats) to reduce steric hindrance between interaction domains and reporter fragments [29].
Frequently Asked Questions (FAQs)
Q1: When should I choose BRET over split-luciferase complementation for my membrane protein study?
A1: BRET is ideal for studying proximity and continuous monitoring of interaction dynamics, as it is reversible and reports on relative distance [31] [36]. Split-luciferase complementation is better for detecting stable interactions and for high-throughput screening, as it generates a cumulative signal but is generally less reversible [30] [29]. For GPCR research, BRET is often preferred for studying receptor activation and trafficking, while complementation assays work well for detecting dimerization events [12] [35].
Q2: What are the key considerations for tagging membrane proteins with luciferase fragments?
A2: Critical factors include: (1) Ensure fragments face the same compartment (cytosolic side for intracellular interactions); (2) Select minimal tags (e.g., 11-aa HiBiT) to reduce steric interference; (3) Verify that tagging does not disrupt membrane localization or function; (4) Test multiple fusion orientations (N- or C-terminal); (5) Include flexible linkers between protein and reporter [29] [33]. For multi-pass membrane proteins, consult structural data or predictive algorithms to identify appropriate terminal positions.
Q3: How can I distinguish specific interaction signals from random collisions?
A3: Several approaches can address this: (1) Perform concentration-dependence studies - specific interactions saturate while random collisions show linear increases; (2) Conduct time-course experiments - specific interactions are stable over time; (3) Include appropriate negative controls with non-interacting partners; (4) Use competition with untagged proteins or known inhibitors; (5) For BRET, calculate the net BRET ratio by subtracting background from negative controls [36] [35].
Q4: What are the advantages of NanoLuc-based systems over traditional luciferases?
A4: NanoLuc offers several advantages: (1) Smaller size (19 kDa) causes less steric interference; (2) Much brighter signal (~150x Renilla or Firefly luciferases); (3) Superior stability (half-life >2 hours); (4) favorable emission spectrum (460 nm) for BRET applications; (5) Compatible with furimazine substrate with glow-type kinetics [31]. These properties make NanoLuc ideal for detecting weak interactions and studying proteins expressed at endogenous levels.
Q5: Can these methods be applied to high-throughput compound screening?
A5: Yes, both technologies are well-suited for high-throughput screening. Split-luciferase complementation is particularly robust for identifying disruptors or enhancers of protein interactions in 384-well formats [34]. BRET systems, especially NanoBRET, enable live-cell screening under physiological conditions and can detect compound effects on interaction kinetics [31] [36]. Both methods generate highly quantifiable data compatible with automated screening platforms.
Essential Research Reagent Solutions
Table 3: Key Reagents for Proximity-Based Luminescence Assays
| Reagent Category | Specific Examples | Function/Application | Technical Notes |
|---|---|---|---|
| Luciferase Donors | NanoLuc, RLuc8, Firefly variants [30] [32] | Energy donor in BRET; complementing fragments | NanoLuc offers brightest signal; Firefly useful for deep-tissue imaging |
| Acceptors/Fluorophores | HaloTag ligands, GFP variants, eYFP [32] [31] | Energy acceptors in BRET | HaloTag compatible with cell-permeable dyes for NanoBRET |
| Substrates | Furimazine, Coelenterazines, D-Luciferin [30] [32] | Luciferase enzyme substrates | Furimazine for NanoLuc (glow-type); Coelenterazine for Renilla (flash-type) |
| Expression Vectors | pcDNA3.1, pLVX, inducible systems [35] | Delivery of fusion constructs | Inducible systems help control expression levels to minimize artifacts |
| Cell Lines | HEK293, HEK293T, HeLa, specialized stable lines [34] [36] [35] | Cellular environment for experiments | Stable lines with endogenous expression reduce overexpression artifacts |
| Detection Instruments | CLARIOStar Plus, Mithras LB 940 [32] [35] | Luminescence measurement | Filter-based readers recommended for optimal BRET sensitivity |
Detailed Experimental Protocols
BRET Assay Protocol for Membrane Protein Interactions
This protocol outlines the steps for measuring protein-protein interactions in mammalian cells using BRET, with specific optimization for membrane proteins [36] [35].
Materials:
- HEK293T or other appropriate cell line
- BRET constructs (NanoLuc-fusion donor and HaloTag-fusion acceptor)
- NanoBRET Nano-Glo Substrate (diluted 1:200) [35]
- White-bottom 96-well plates
- CLARIOStar Plus or similar microplate reader with appropriate filter sets
Procedure:
- Cell seeding: Harvest and resuspend cells in assay media. Seed 50,000-100,000 cells per well in white-bottom 96-well plates and incub overnight [36] [35].
- Transfection: Transfect with BRET constructs using appropriate method. Maintain donor:acceptor DNA ratio between 1:3 and 1:10 for optimal signal [36].
- Substrate addition: Add NanoBRET Nano-Glo Substrate (final dilution 1:200) 15 minutes prior to luminescence measurement [35].
- Luminescence measurement: Read luminescence using 460±40 nm (donor) and 615±20 nm (acceptor) filters at 37°C [35].
- Data analysis: Calculate BRET ratio as (acceptor emission / donor emission). Normalize to background from donor-only controls [35].
Split-Luciferase Complementation Assay Protocol
This protocol describes the implementation of split-luciferase complementation assays to detect specific protein-protein interactions, optimized for transmembrane proteins [34] [33].
Materials:
- HEK293T cells
- Expression plasmids encoding protein fusions with luciferase fragments
- Lipid-based transfection reagent
- Serum-free medium
- Furimazine substrate solution
- Luminescence microplate reader
Procedure:
- Cell preparation: Plate adherent HEK293T cells onto clear bottom, white side 96-well plate and culture overnight [33].
- Transfection: Dilute plasmids in serum-free medium to 6.25 ng/μL for each construct. Add lipid-based transfection reagent at appropriate lipid-to-DNA ratio. Incubate according to manufacturer's instructions [33].
- Transfection mixture addition: Add 8 μL of lipid-DNA mixture to designated wells. Culture cells for 20-24 hours [33].
- Medium replacement: Replace conditioned medium with 100 μL serum-free medium, taking care not to detach cells [33].
- Substrate preparation and addition: Mix one volume furimazine with 19 volumes dilution buffer. Add 25 μL furimazine working solution to each well [33].
- Luminescence measurement: Equilibrate plate at 37°C for 10-15 minutes in microplate reader. Read luminescence with 0.3-second integration time [33].
BRET and split-luciferase complementation assays represent powerful methodologies that have transformed our ability to study membrane protein interactions in live cells under physiological conditions. By providing high sensitivity, temporal resolution, and compatibility with high-throughput formats, these techniques address critical limitations in membrane protein research. The troubleshooting guidelines and protocols presented here offer researchers practical tools to overcome common experimental challenges, thereby enhancing data reliability and accelerating discovery in drug development and basic membrane biology.
The investigation of membrane protein interactions presents a significant challenge in molecular biology. Traditional techniques like Yeast Two-Hybrid (Y2H) and Co-Immunoprecipitation (Co-IP) are limited by their inability to capture transient interactions in living cells and often generate false positives for membrane-associated complexes [37]. Förster Resonance Energy Transfer (FRET) microscopy overcomes these limitations by enabling the detection of dynamic interactions with a spatial resolution of 1-10 nanometers, functioning as a sensitive "molecular ruler" within live cells [37] [38].
This technical resource center focuses on three advanced FRET modalities—FLIM-FRET, smFRET, and TR-FRET—which provide robust solutions for quantifying protein interactions and conformational changes in real time. These techniques are particularly vital for drug development, as they can characterize the mechanisms and efficacy of therapeutic compounds designed to modulate protein interactions [37].
FRET Technique Comparison and Selection Guide
Quantitative Comparison of FRET Modalities
The following table summarizes the key operational characteristics and ideal use cases for each advanced FRET technique, helping researchers select the most appropriate method for their experimental goals.
Table 1: Technical Overview of Advanced FRET Methods
| Technique | Key Readout | Spatial Resolution | Key Advantage | Primary Application |
|---|---|---|---|---|
| FLIM-FRET | Donor fluorescence lifetime [39] [40] | <10 nm [40] | Independent of fluorophore concentration; robust to expression level variations [40] | Quantifying protein-protein interactions and topology in live cells [40] |
| smFRET | FRET efficiency from single molecules [41] | ~2 Å precision, ~5 Å accuracy [41] | Reveals population heterogeneity and detects transient conformational dynamics [41] | Characterizing structural dynamics and distances in proteins; identifying subpopulations [41] |
| TR-FRET | Time-resolved fluorescence lifetime [37] | Nanometer scale [37] | Eliminates background fluorescence via time-gated detection; ideal for high-throughput screening [37] | Screening for small-molecule modulators of protein-protein interactions [37] |
Visual Guide to FRET Techniques and Their Applications
The diagram below illustrates the core workflow and primary application of each FRET technique within the context of a live-cell experiment.
Troubleshooting Guides and FAQs
This section addresses common experimental challenges and provides targeted solutions to ensure robust and reproducible FRET data.
FLIM-FRET Troubleshooting
- Problem: Low signal-to-noise ratio in lifetime images.
- Solution: Implement two-photon microscopy with near-infrared light (~800-950 nm) to reduce cellular autofluorescence and minimize photodamage in plant or tissue samples [40].
- Problem: Apparent lifetime changes not due to FRET.
- Solution: Always include a donor-only control (e.g., cells expressing only the donor fluorophore) to establish the baseline fluorescence lifetime (τD) in the absence of an acceptor [40].
- Problem: Uncertainty in data interpretation due to environmental effects.
- Solution: Note that the fluorescence lifetime is largely independent of local environmental factors like pH, making FLIM-FRET more robust than intensity-based methods for quantitative analysis [40].
smFRET Troubleshooting
- Problem: Low reproducibility of FRET efficiency histograms between labs.
- Solution: Implement rigorous calibration routines. A recent multi-laboratory study demonstrated that using standardized controls and analysis yields a high interdye distance precision of ≤2 Å [41].
- Protocol: Use alternating-laser excitation (ALEX/PIE) to correct for spectral crosstalk, determine γ-factor for detection differences, and exclude molecules with inactive acceptors [41].
- Problem: Inability to detect structural dynamics.
- Solution: Refine data analysis to identify subpopulations within FRET efficiency histograms. smFRET can reliably detect distance fluctuations as small as 5 Å on sub-millisecond timescales [41].
- Problem: Dye perturbations affecting protein function.
- Solution: Validate that site-specific dye labeling (e.g., via cysteine mutations) does not impair protein function using a complementary activity assay before performing FRET experiments [41].
General FRET & Calibration Troubleshooting
- Problem: FRET ratio is sensitive to imaging parameters and cannot be compared across sessions.
- Solution: Use internal calibration standards. Integrate "FRET-ON" and "FRET-OFF" standard constructs into your experiment. The fluorescence signals from these standards can be used to normalize the FRET ratio, compensating for variability in laser power and detector sensitivity [42].
- Problem: Photobleaching obscures true FRET changes during long-term imaging.
- Solution: The calibration strategy using FRET standards also corrects for signal drifts caused by photobleaching, restoring the reciprocal relationship between donor and acceptor signals and validating biosensor responses over time [42].
- Problem: Difficulty multiplexing multiple FRET biosensors.
- Solution: Employ a biosensor barcoding method. Label cells expressing different biosensors with distinct pairs of blue or red fluorescent proteins targeted to different subcellular locations. A machine learning model can then identify each biosensor's identity based on its barcode pattern during imaging [42].
Detailed Experimental Protocols
FRET-FLIM Protocol for Protein-Protein Interaction in Plant Cells
This protocol is adapted for investigating membrane protein complexes in tobacco leaf epidermal cells [40].
- Step 1: Construct Generation. Fuse your proteins of interest (e.g., membrane subunits A and B) to selected donor (e.g., eGFP) and acceptor (e.g., mRFP) fluorophores via standard molecular cloning.
- Step 2: Transient Expression.
- Transform the constructs into Agrobacterium strains.
- Grow bacterial cultures in YEB medium with appropriate antibiotics to an OD600 of ~0.8 [40].
- Centrifuge and resuspend the cells in infiltration buffer (containing acetosyringone to facilitate infection) [40].
- Infiltrate the suspensions into the abaxial side of 4-6 week-old Nicotiana benthamiana leaves using a needleless syringe [40].
- Step 3: Incubation. Incubate infiltrated plants for 48-72 hours under standard growth conditions to allow for high-level protein expression [40].
- Step 4: Data Acquisition.
- Mount a small section of the expressing leaf epidermis on a microscope slide.
- Using a confocal microscope equipped with a TCSPC module and a pulsed laser (e.g., a Ti:Sapphire laser for two-photon excitation at 950 nm for eGFP), acquire FLIM data by collecting donor emission (500-550 nm) only [40].
- First, image cells expressing the donor-only construct to determine the reference lifetime (τD).
- Then, image cells co-expressing the donor and acceptor fusion proteins (τDA).
- Step 5: Data Analysis.
- Fit the fluorescence decay curves to a multi-exponential model to calculate the average fluorescence lifetime.
- Calculate the FRET efficiency (E) using the formula: E = 1 - (τDA / τD) [40]
- A significant decrease in the donor lifetime in the presence of the acceptor indicates a positive interaction.
smFRET Protocol for Characterizing Protein Conformational Dynamics
This protocol outlines the key steps for a confocal smFRET study to probe structural dynamics, as validated in a multi-laboratory study [41].
- Step 1: Sample Preparation.
- Labeling: Introduce two cysteine mutations at specific sites on the protein surface for dye conjugation. Purify the protein and label it stochastically with a donor (e.g., Alexa Fluor 546) and an acceptor (e.g., Alexa Fluor 647) dye. Remove free dye [41].
- Functionality Check: Validate the activity of the labeled protein using a functional assay (e.g., ligand binding via microscale thermophoresis) before FRET measurements [41].
- Measurement Setup: Dilute the labeled protein to picomolar concentration in imaging buffer to ensure the detection of single molecules. Use a confocal microscope equipped with alternating laser excitation (ALEX or PIE) [41].
- Step 2: Data Collection.
- Collect photons from single molecules diffusing through the confocal volume.
- Record signals in donor and acceptor channels during both donor and acceptor excitation cycles [41].
- Step 3: Data Correction and Analysis.
- Correction Factors: Determine the correction factors (α, β, γ, δ) for spectral crosstalk, direct excitation, and detection efficiency differences using donor-only and acceptor-only samples [41].
- FRET Efficiency: Calculate the accurate, proximity-dependent FRET efficiency (E) for each burst of photons from a single molecule.
- Histograms: Plot E histograms to visualize the distribution of conformational states. The presence of multiple peaks indicates population heterogeneity [41].
- Distance Calculation: Convert FRET efficiencies to inter-dye distances, achieving a precision of ≤2 Å and an accuracy of ≤5 Å for well-behaved systems [41].
The Scientist's Toolkit: Essential Research Reagents
The following table lists critical reagents and their functions for successfully implementing advanced FRET experiments.
Table 2: Key Research Reagent Solutions for FRET Experiments
| Reagent / Material | Function / Explanation | Example Use Case |
|---|---|---|
| eGFP & mRFP Pair | Donor and acceptor fluorescent proteins for FP-FRET. Spectral overlap allows efficient energy transfer [40]. | FRET-FLIM interaction studies in live plant cells [40]. |
| Alexa Fluor 546 & Alexa Fluor 647 | Organic dyes for smFRET. High brightness and photostability are essential for single-molecule detection [41]. | Site-specific labeling of protein cysteine mutants for confocal smFRET [41]. |
| FRET Calibration Standards | Genetically encoded constructs locked in "FRET-ON" and "FRET-OFF" states [42]. | Normalizing FRET ratios to correct for day-to-day instrument variability [42]. |
| Cysteine Variants | Engineered proteins with cysteine residues at specific sites for covalent, site-specific attachment of maleimide-functionalized dyes [41]. | Ensuring defined dye labeling positions for accurate distance measurements [41]. |
| Agrobacterium tumefaciens | A bacterial vector used for transient transformation and high-level protein expression in plant leaves [40]. | Delivering FRET biosensor genes into tobacco leaf epidermal cells [40]. |
| Acceptor-only & Donor-only Samples | Control samples expressing only the acceptor or donor fluorophore [42] [41]. | Essential for determining spectral crosstalk and calculating accurate FRET efficiencies [42] [41]. |
Technical Support & Troubleshooting Center
This resource is designed to support researchers utilizing nanodisc and styrene-maleic acid (SMA) copolymer technologies for structural studies of membrane proteins via cryo-EM, within the broader thesis of overcoming limitations in native membrane protein interaction detection.
Frequently Asked Questions (FAQs) & Troubleshooting Guides
FAQ 1: Why might my membrane protein show constitutive activity or altered function after SMA copolymer extraction?
- Issue: Following extraction with SMA copolymer from unstimulated cells, receptors like the insulin receptor (InsR) exhibit autophosphorylation in the absence of ligand, which is not observed with traditional detergent extraction [43].
- Potential Cause & Solution: The insertion of the SMA polymer into the membrane bilayer during the extraction process may itself perturb the local membrane environment or protein conformation, mimicking or inducing an activated state [43]. This highlights a critical consideration for functional studies.
- Troubleshooting Steps:
- Functional Validation is Mandatory: Always couple structural extraction with a functional assay specific to your protein to confirm native behavior is retained [43] [44].
- Titrate Polymer Ratio: Systematically vary the SMA-to-membrane ratio. Excess polymer can interfere with protein dynamics and activation, as seen with rhodopsin [44]. Find the minimal effective concentration for solubilization.
- Consider Alternative Copolymers: Newer copolymers like diisobutylene/maleic acid (DIBMA) or SMA-QA variants may offer a less perturbing extraction profile for sensitive proteins [43].
- Troubleshooting Steps:
FAQ 2: How can I optimize nanodisc homogeneity for high-resolution cryo-EM, especially with smaller bitopic membrane proteins?
- Issue: When the molecular weight of the target transmembrane protein is similar to that of the nanodisc itself, imperfections in the nanodisc preparation can dominate the cryo-EM signal and limit resolution [45].
- Potential Cause & Solution: Sample heterogeneity arising from variable lipid-to-scaffold protein (MSP) ratios, incomplete detergent removal, or non-uniform protein incorporation.
- Troubleshooting Steps:
- Pre-form and Characterize Empty Nanodiscs: First prepare and purify bare nanodiscs of your chosen size (using MSP1D1, MSP1E3D1, etc.) to ensure a monodisperse starting population [45].
- Optimize Reconstitution Stoichiometry: Carefully optimize the molar ratios of membrane protein, lipids, and MSP. This is critical when the protein occupies a small fraction of the disc [45].
- Employ Orthogonal Quality Controls: Use size-exclusion chromatography (SEC) coupled with multi-angle light scattering (MALS) or mass photometry to assess sample homogeneity and oligomeric state accurately and rapidly before proceeding to cryo-EM grid preparation [13].
- Troubleshooting Steps:
FAQ 3: My SMA-solubilized sample precipitates. What are the common causes?
- Issue: Precipitation of SMA nanodiscs (SMALPs) during purification or buffer exchange.
- Potential Causes & Solutions:
- Divalent Cations: SMA copolymers are sensitive to divalent cations like Mg²⁺ or Ca²⁺, which cause disassembly and precipitation [43]. Solution: Use chelating agents (e.g., EDTA) in all buffers and ensure water purity.
- Low pH: SMA precipitates at low pH when the maleic acid groups become protonated [43]. Solution: Maintain buffer pH above 6.5, typically at 7.4-8.0.
- Polymer Instability: Commercial SMA is polydisperse. Solution: Consider using more homogeneous RAFT-synthesized SMA or alternative copolymers like DIBMA for improved stability [43].
FAQ 4: How do I choose between MSP Nanodiscs and SMA Copolymers (SMALPs) for my project?
- Decision Factors: This choice depends on the experimental goal within the context of studying native interactions.
- Choose SMA/SMALPs for: Direct, detergent-free extraction from native membranes [46]; preserving a native lipid annulus [47]; studying protein-lipid interactions from a complex source.
- Choose MSP Nanodiscs for: Full control over lipid composition during reconstitution [45]; working with proteins that require an initial detergent solubilization step [48]; achieving highly monodisperse, homogeneous samples critical for high-resolution cryo-EM [45].
Table 1: Comparison of Membrane Protein Solubilization Methods
| Method | Key Advantage | Key Limitation | Typical Size Range | Native Lipids Retained? |
|---|---|---|---|---|
| Detergent Micelles | Wide screening available, simple [46] | Poor bilayer mimic, destabilizing [46] [13] | 5-10 nm [43] | No |
| MSP Nanodiscs | Controlled lipid environment, stable [46] [45] | Requires detergent step, non-native extraction [48] | ~10 nm (tunable) [46] [45] | No (defined) |
| SMA Copolymers (SMALPs) | Direct native extraction, retains native lipids [46] [47] | Sensitive to pH/cations, may alter function [43] [44] | 10-30 nm [43] | Yes |
| Amphipols | High stability, low exchange rate [46] | Does not provide a lipid bilayer [46] | Variable | No |
Table 2: Functional Outcomes in SMA-Extracted Proteins (Case Studies)
| Protein (System) | Observation Post-SMA Extraction | Suggested Cause | Reference |
|---|---|---|---|
| Insulin Receptor (3T3L1 cells) | Constitutive autophosphorylation & IRS1 phosphorylation. No response to added insulin. | Polymer insertion may perturb membrane, inducing activation. Distal signaling (Akt) not activated. | [43] |
| Rhodopsin (Model membranes) | High SMA/protein ratio solubilizes more protein but blocks formation of active Meta II state. | Excess polymer interferes with conformational dynamics required for activation. | [44] |
| Various (GPCRs, transporters) | Successful ligand binding and effector recruitment demonstrated. | Correct folding and function can be preserved under optimized conditions. | [43] |
Detailed Experimental Protocols
Protocol 1: Reconstitution of Bitopic Membrane Proteins into MSP Nanodiscs for Cryo-EM Objective: To generate homogeneous, monodisperse complexes of a single-span membrane protein in nanodiscs [45].
- Preparation of Components:
- Lipids: Dry down chloroform-solubilized lipids (e.g., POPC) under nitrogen gas and solubilize in lipid solubilization buffer (20 mM Tris pH 7.4, 100 mM NaCl, 0.5 mM EDTA, 70 mM sodium cholate) [45].
- Membrane Scaffold Protein (MSP): Express and purify MSP (e.g., MSP1D1 for ~9.5 nm discs) using standard IMAC protocols [45].
- Target Membrane Protein: Purify the detergent-solubilized protein (e.g., in DDM) to monodispersity.
- Reconstitution:
- Mix detergent-solubilized protein with solubilized lipids at an optimized molar ratio.
- Add MSP at a molar ratio typically 2-4x the lipid concentration.
- Initiate self-assembly by removing detergent via dialysis or incubation with Bio-Beads SM-2 adsorbents [45].
- Purification & Validation:
- Purify the reconstituted complex using size-exclusion chromatography (SEC) in a compatible buffer (e.g., 20 mM Tris pH 7.4, 100 mM NaCl, 0.5 mM EDTA).
- Validate homogeneity using SEC-MALS, negative stain EM, and mass photometry to confirm correct oligomerization within discs [13].
Protocol 2: Direct Extraction of Membrane Proteins Using SMA Copolymer Objective: To solubilize membrane proteins directly from cellular membranes while preserving a native lipid environment [46] [43].
- Cell Membrane Preparation: Harvest cells (e.g., 3T3L1 fibroblasts) and prepare a membrane pellet or use intact cells.
- Extraction:
- Resuspend membranes/cells in a divalent-cation-free buffer (e.g., PBS, pH 7.4).
- Add a 2:1 or 3:1 styrene:maleic acid copolymer (e.g., SMA2000) from a stock solution (e.g., 5% w/v in water). The optimal polymer-to-lipid ratio must be determined empirically [43] [44].
- Incubate with gentle agitation for 1-3 hours at room temperature or 37°C.
- Isolation of SMALPs:
- Remove insoluble material by ultracentrifugation (e.g., 100,000 x g, 1 hour, 4°C).
- The supernatant contains the solubilized SMALPs.
- For further purification, the supernatant can be subjected to affinity chromatography (if the target protein is tagged) or SEC.
- Critical Control: Always perform a parallel extraction with a standard detergent (e.g., in RIPA buffer) for functional and compositional comparison [43].
Visualizations
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Nanodisc & SMA-Based Cryo-EM Studies
| Reagent Category | Specific Examples | Primary Function & Consideration |
|---|---|---|
| Membrane Scaffold Proteins (MSPs) | MSP1D1 (~9.5 nm), MSP1E3D1 (~12 nm), MSP2N2 (~15 nm) [45] | Forms the protein belt around nanodiscs, controlling disc size. Choice dictates the diameter of the lipid bilayer patch. |
| Lipids | POPC, DOPC, native lipid extracts [45] | Forms the bilayer environment within nanodiscs. Synthetic lipids allow control; native extracts preserve natural composition. |
| Styrene-Maleic Acid (SMA) Copolymers | SMA2000 (2:1), SMA3000 (3:1), RAFT-SMA, DIBMA [43] | Directly solubilizes membranes into SMALPs. Ratio affects properties; new variants (RAFT, DIBMA) offer homogeneity and stability. |
| Detergents (for MSP reconstitution) | DDM, Triton X-100, Sodium Cholate [45] | Initial solubilization of membrane proteins prior to MSP reconstitution. Critical for the detergent removal step. |
| Detergent Removal Agents | Bio-Beads SM-2 Adsorbent [45] | Hydrophobic beads that absorb detergent monomers, driving nanodisc self-assembly during dialysis or incubation. |
| Buffers & Additives | Tris/HEPES buffers, NaCl, EDTA, Protease inhibitors [45] | Maintain pH and ionic strength. EDTA is critical for SMA stability to chelate divalent cations. |
| Quality Control Tools | Mass Photometer, SEC-MALS system, Negative Stain EM [13] | Mass photometry is highlighted as a rapid, single-molecule method to assess sample homogeneity, oligomerization, and complex integrity before cryo-EM [13]. |
Protein-protein interactions (PPIs) are fundamental regulators of most biological processes, including signal transduction, cell cycle regulation, and metabolic pathways [49]. The deregulation of PPIs can lead to serious diseases such as cancer, autoimmune disorders, and neurodegenerative conditions [50]. Traditionally, PPIs have been identified using experimental methods like yeast two-hybrid screening, co-immunoprecipitation, and mass spectrometry [49]. However, these techniques are often time-consuming, resource-intensive, and challenging to scale [49] [50].
The field is undergoing a transformative shift with the inclusion of deep learning, a cornerstone of artificial intelligence known for its remarkable pattern recognition capabilities [49]. Unlike conventional machine learning algorithms that rely on manually engineered features, deep learning can autonomously extract meaningful features from complex biological data, making it particularly well-suited for processing large-scale PPI datasets [49]. This capability is crucial for membrane protein research, where traditional experimental methods face significant hurdles due to issues with protein solubility, expression, and the complex cellular environment [51] [52].
Core Deep Learning Architectures for PPI Prediction
Deep learning encompasses several neural network architectures, each with distinct advantages for processing different types of biological data.
Graph Neural Networks (GNNs)
GNNs operate on graph structures and use message-passing mechanisms to adeptly capture local patterns and global relationships in protein structures [49]. By aggregating information from neighboring nodes, GNNs generate node representations that reveal complex interactions and spatial dependencies [49]. Key variants include:
- Graph Convolutional Networks (GCNs): Employ convolutional operations to aggregate information from neighboring nodes [49].
- Graph Attention Networks (GATs): Introduce an attention mechanism that adaptively weights neighboring nodes based on their relevance [49].
- GraphSAGE: Designed for large-scale graph processing, utilizing neighbor sampling and feature aggregation to reduce computational complexity [49].
- Graph Autoencoders (GAE): Utilize an encoder-decoder framework to generate compact, low-dimensional node embeddings for graph reconstruction or predictive tasks [49].
Researchers have developed innovative frameworks that integrate these approaches. For instance, the AG-GATCN framework integrates GAT and temporal convolutional networks to provide robust solutions against noise interference in PPI analysis [49]. The RGCNPPIS system integrates GCN and GraphSAGE to simultaneously extract macro-scale topological patterns and micro-scale structural motifs [49].
Convolutional and Recurrent Neural Networks
- Convolutional Neural Networks (CNNs): Particularly effective for processing spatial hierarchies in data. In PPI prediction, 1D-CNNs can extract local sequence motifs and patterns from protein sequences [50]. The Deep_PPI model uses two 1D-CNN branches to process protein pairs, with outputs concatenated by a fully connected layer [50].
- Long Short-Term Memory Networks (LSTMs): A type of recurrent neural network capable of learning long-range dependencies in sequential data, making them suitable for analyzing amino acid sequences where distant residues might influence interaction interfaces [51].
Multi-Task Learning Frameworks
Multi-task learning allows simultaneous prediction of multiple related properties, improving generalizability. The Membrane Association and Secondary Structure Predictor (MASSP) integrates a multi-layer 2D-CNN with an LSTM network to predict both residue-level structural attributes and protein-level structural classes [51]. This approach enables automatic distinction between different protein classes (bitopic, α-helical, β-barrel, and soluble) while identifying transmembrane segments and topologies when present [51].
Emerging Architectures
Recent approaches have incorporated attention-driven Transformers, multimodal integration of sequence and structural data, and transfer learning via language models like BERT and ESM [49]. These advancements address challenges such as data imbalances, variations, and high-dimensional feature sparsity in PPI prediction [49].
Experimental Protocols and Data Processing
Standardized Experimental Workflow
A typical deep learning workflow for PPI prediction involves several key stages, from data collection to model validation. The following diagram illustrates this process:
Feature Encoding and Data Preparation
Proper feature representation is crucial for transforming biological protein sequences into numerical feature vectors that deep learning models can process [50]. The table below summarizes common feature encoding strategies:
Table 1: Feature Encoding Strategies for PPI Prediction
| Encoding Method | Description | Application Example | Advantages |
|---|---|---|---|
| One-Hot Encoding | Represents 20 native amino acids and one special residue using a 21D vector, then applies Keras one-hot function [50]. | Deep_PPI model for multiple species PPI prediction [50]. | Simple implementation, avoids manual feature engineering. |
| Binary Profile with PaddVal | Uses tokenization and zero-padding to ensure uniform sequence length across protein pairs [50]. | Standardization of input dimensions for CNN-based models [50]. | Handles variable-length sequences, compatible with standard neural networks. |
| Position-Specific Scoring Matrix (PSSM) | Contains log-odds of each amino acid observed at sequence positions over evolutionary timescale [51]. | MASSP model for predicting membrane association and secondary structure [51]. | Captures evolutionary conservation information. |
| Docking Site Motif Analysis | Identifies specific interaction motifs like D-sites (R/K₁₋₃-X₁₋₆-φ-X-φ) in MAPK phosphatases [53]. | Predicting PPIs between MAPKs and PP2Cs in B. distachyon [53]. | Incorporates structural biology insights into sequence analysis. |
For the binary profile encoding with PaddVal strategy, researchers typically set the PaddVal value to the length of 90% of protein sequences, which has been experimentally verified to perform better than other values (75%, 80%, 85%, 95%) [50]. This approach adds zeros if a protein's length is less than the PaddVal value and truncates sequences longer than this value [50].
Benchmark Datasets for PPI Prediction
High-quality benchmark datasets are essential for training and evaluating deep learning models. The table below summarizes key databases used in PPI prediction research:
Table 2: Key Databases for PPI Prediction Research
| Database Name | Description | Application in PPI Research |
|---|---|---|
| STRING | Database of known and predicted protein-protein interactions across various species [49]. | Provides interaction data for training and validation of prediction models. |
| BioGRID | Database of protein-protein and gene-gene interactions from various species [49]. | Source of experimentally verified interactions for benchmark datasets. |
| DIP | Database of experimentally verified protein-protein interactions [49]. | Curated source of positive interaction examples for model training. |
| OPM | Database of orientational parameters of membranes in the PDB [51]. | Source of membrane protein structures and topological data. |
| HPRD | Human protein reference database with interaction, enzymatic, and cellular localization data [49]. | Primary source for human PPI data in species-specific prediction. |
When constructing benchmark datasets, researchers typically prune sequences to 25% sequence identity using servers like PISCES to remove redundancy [51]. Datasets are often split with ratios of 8:1:1 for training, validation, and testing while maintaining constant fractions of each protein class in each subset [51].
Troubleshooting Common Computational Challenges
Data Quality and Availability Issues
Problem: Limited or Imbalanced Training Data
- Symptoms: Model exhibits poor generalization, low accuracy on independent test sets, or bias toward majority classes.
- Solutions:
- Apply data augmentation techniques such as random sequence cropping, amino acid substitution based on similarity matrices, or synthetic minority over-sampling.
- Utilize transfer learning by pre-training on large protein language models (e.g., ESM, ProtBERT) followed by fine-tuning on specific PPI tasks [49].
- Incorporate multi-task learning to leverage related predictive tasks and improve model robustness [51].
Problem: Inconsistent Feature Representation
- Symptoms: Model fails to converge or shows erratic performance during training.
- Solutions:
- Implement standardized preprocessing pipelines with consistent sequence padding and normalization strategies [50].
- For membrane proteins, incorporate structural class prediction as an auxiliary task to guide feature learning [51].
- Use ensemble methods that combine multiple feature representations (sequence, evolutionary, structural) [51].
Model Performance and Generalization Problems
Problem: Poor Generalization to Novel Protein Classes
- Symptoms: High performance on validation data but significant drop on external test sets, particularly with proteins dissimilar to training examples.
- Solutions:
- Implement rigorous cross-validation strategies that account for sequence similarity between training and test sets [50].
- For membrane proteins specifically, ensure training data includes diverse topological classes (bitopic, α-helical, β-barrel) [51].
- Regularize models using dropout, weight decay, or early stopping to prevent overfitting [50].
Problem: Physically Implausible Predictions
- Symptoms: Models generate accurate binding poses but with steric clashes, incorrect bond geometries, or unrealistic interaction patterns.
- Solutions:
- Incorporate physical constraints into loss functions or use hybrid approaches that combine deep learning with physics-based scoring [54].
- Implement post-processing validation with tools like PoseBusters to check chemical and geometric consistency [54].
- For docking applications, prefer generative diffusion models or hybrid methods over pure regression-based approaches, which tend to produce more physically valid poses [54].
Membrane Protein-Specific Challenges
Problem: Difficulty Predicting Transmembrane Topology
- Symptoms: Inaccurate identification of membrane-spanning segments or incorrect orientation relative to the membrane.
- Solutions:
- Implement multi-task frameworks like MASSP that simultaneously predict secondary structure, location, orientation, and topology [51].
- Incorporate lipid accessibility constraints and positive-inside rule priors during model training [51].
- Use ensembles of specialized predictors for different membrane protein classes (α-helical vs. β-barrel) [51].
Problem: Handling Peripheral Membrane Protein Interactions
- Symptoms: Failure to detect transient interactions or membrane-associated complexes.
- Solutions:
- Apply methodologies like z-scan fluorescence fluctuation spectroscopy that can disentangle fluorescence contributions from membrane-bound and cytoplasmic pools [55].
- Develop models that explicitly account for reversible membrane association-dissociation kinetics [55].
- Incorporate spatial temporal features from live-cell imaging data when available [52].
Table 3: Essential Resources for Computational PPI Research
| Resource Category | Specific Tools/Methods | Function/Purpose |
|---|---|---|
| Experimental Validation Methods | ReLo (Relocalization Assay) [52] | Simple, rapid cell culture-based method for detecting direct binary PPIs, especially useful for large or insoluble proteins. |
| Experimental Validation Methods | Co-immunoprecipitation (Co-IP) [56] | Biochemical method for confirming physical interactions between proteins; requires careful control experiments. |
| Experimental Validation Methods | Yeast Two-Hybrid (Y2H) [57] | Genetic system for detecting PPIs; prone to false positives requiring rigorous validation. |
| Experimental Validation Methods | Z-scan Fluorescence Fluctuation Spectroscopy [55] | Technique for determining oligomeric state of peripheral membrane proteins in cytoplasm and at plasma membrane. |
| Computational Frameworks | Deep_PPI [50] | 1D-CNN architecture for predicting PPIs from sequence alone using tokenization and one-hot encoding. |
| Computational Frameworks | MASSP [51] | Multi-task deep learning method integrating 2D-CNN and LSTM for predicting membrane association and secondary structure. |
| Computational Frameworks | AG-GATCN [49] | Graph neural network framework combining graph attention networks and temporal convolutional networks. |
| Validation Tools | PoseBusters [54] | Toolkit for evaluating docking predictions against chemical and geometric consistency criteria. |
| Validation Tools | Docking Energy Calculations [53] | In silico approach for predicting interaction stability based on global energy minimization. |
Validation Frameworks and Integration with Experimental Methods
Computational predictions require rigorous validation to ensure biological relevance. The following diagram illustrates an integrated validation workflow for computational predictions:
Physical Plausibility Assessment
Before experimental validation, computational predictions should be evaluated for physical plausibility:
- Steric Clash Detection: Use tools like PoseBusters to identify physically impossible atomic overlaps [54].
- Geometric Consistency: Validate bond lengths, angles, and stereochemistry against known structural data [54].
- Interaction Pattern Analysis: Ensure predicted interfaces recapitulate known interaction motifs (e.g., D-sites in MAPK phosphatases) [53].
Experimental Validation Techniques
- ReLo Assay: Particularly valuable for membrane and large proteins, this live-cell imaging approach detects direct binary interactions through protein relocalization [52]. The method works by fusing one protein to a membrane-anchoring domain and monitoring colocalization of its potential partner [52].
- Crosslinking Strategies: For transient interactions, membrane-permeable crosslinkers like DSS can "freeze" interactions inside cells, while membrane-impermeable crosslinkers like BS3 work on the cell surface [57].
- Co-IP Optimization: Use gentle lysis buffers without ionic detergents like sodium deoxycholate to preserve native interactions [56]. Include appropriate controls: bead-only, isotype, and input lysate controls are essential for interpreting results [56].
Functional Validation in Biological Context
- Mutational Analysis: Introduce point mutations at predicted interaction interfaces and assess impact using ReLo or other interaction assays [52].
- Domain Mapping: Systematically test protein domains and truncations to identify minimal interaction regions [52].
- Pharmacological Modulation: Test whether small molecules or drugs can disrupt or enhance predicted interactions, providing functional validation and potential therapeutic insights [52].
Future Perspectives and Concluding Remarks
Deep learning approaches for PPI prediction are rapidly evolving, with several emerging trends shaping the future of the field. Multi-modal architectures that integrate sequence, structural, and evolutionary information are showing improved performance across diverse protein classes [49]. For membrane proteins specifically, multi-task frameworks that simultaneously predict multiple structural attributes offer particular promise [51].
Despite significant progress, challenges remain in predicting transient interactions, condition-specific PPIs, and interactions involving intrinsically disordered regions [49]. Furthermore, most current methods exhibit limited generalization to novel protein folds or families not represented in training data [54]. Addressing these limitations will require both algorithmic innovations and expanded, curated datasets that capture the dynamic nature of protein interactions.
For researchers focusing on membrane proteins, the integration of computational predictions with specialized experimental assays like ReLo and z-scan FFS provides a powerful approach for tackling the unique challenges of this protein class [55] [52]. As deep learning methodologies continue to mature, they hold increasing potential to illuminate the complex interaction networks that underlie cellular function and dysfunction, ultimately accelerating drug discovery and therapeutic development.
Surface Plasmon Resonance (SPR) and Biolayer Interferometry (BLI) are two dominant optical techniques for label-free, real-time analysis of biomolecular interactions. SPR measures changes in the refractive index on a thin metal film to monitor binding events, while BLI analyzes shifts in interference patterns to measure changes in the thickness of a molecular layer on a biosensor tip [58] [59] [60]. For researchers studying membrane proteins—a class of targets notoriously difficult to characterize due to their hydrophobic nature and instability outside native membrane environments—these technologies provide a critical means to quantify interactions with lipids, drugs, and other proteins without the need for fluorescent labels that can alter protein function [13].
This technical support center addresses the specific experimental challenges you may encounter when applying SPR and BLI to your research, providing targeted troubleshooting guides and FAQs to ensure data reliability.
The following table summarizes the core principles and characteristics of each technology.
Table 1: Comparison of SPR and BLI Technologies
| Feature | Surface Plasmon Resonance (SPR) | Biolayer Interferometry (BLI) |
|---|---|---|
| Core Principle | Measures refractive index changes via resonance angle shift on a gold film [58] [59] | Measures thickness changes of biomolecular layers via interference pattern shifts [61] [60] |
| Assay Format | Continuous flow in microfluidic channels [59] | "Dip-and-read" format in microplates [61] [60] |
| Key Components | Gold-coated sensor chip, microfluidic system, optical prism [58] | Disposable fiber-optic biosensor [61] |
| Sensitivity | High (suitable for low-concentration samples and detailed kinetics) [62] | Moderate (suited for medium/high concentrations and rapid screening) [62] |
| Throughput | Moderate (depends on number of flow cells) [58] | High (supports 96- or 384-well plates) [61] [60] |
| Data Output | Real-time binding curves for determining association rate ((k{on})), dissociation rate ((k{off})), and affinity ((K_D)) [58] [59] | Real-time binding curves for determining (k{on}), (k{off}), and (K_D) [61] [59] |
| Primary Limitation | Requires immobilization; potential for surface artifacts and channel clogging [63] | Requires immobilization; potential for sample evaporation and lower kinetic resolution [59] [63] |
Experimental Workflow Diagrams
Title: SPR Experimental Workflow
Title: BLI Experimental Workflow
SPR Troubleshooting Guide
Frequently Asked Questions
Q1: My sensorgram shows a high response during the baseline, or a "drift" effect. What is the cause? This is often due to non-specific binding (NSB) where analytes stick to the sensor surface non-specifically [63]. For membrane proteins in detergents, the detergent itself can cause NSB. To resolve this:
- Optimize your running buffer: Increase ionic strength or include a mild detergent [63].
- Include a blocking step: Use inert proteins like BSA or casein to block non-specific sites on the sensor surface [63].
- Use a different chip chemistry: Switch to a sensor surface with different properties to reduce hydrophobic or electrostatic interactions.
Q2: I get inconsistent binding kinetics between replicates. How can I improve reproducibility? This can stem from several factors related to the immobilization stability and sample handling:
- Ensure uniform ligand immobilization: Use a consistent coupling protocol and confirm similar immobilization levels (Response Units, RU) across flow cells [58].
- Check for sample aggregation: Centrifuge or filter your membrane protein samples prior to injection to remove aggregates that can clog the microfluidic system [13].
- Verify accurate analyte concentration: Use a fresh, validated method to quantify your protein concentration, especially after purification in detergents or amphipols [63].
Q3: The sensor surface cannot be regenerated effectively between cycles. What should I do? Harsh regeneration conditions can damage the immobilized ligand, while mild conditions fail to remove bound analyte [59].
- Perform a regeneration scouting experiment: Test a series of buffers (e.g., low pH, high salt, mild detergent) on a single flow cell to find the minimal effective condition [59].
- Consider a more tolerant immobilization chemistry: If your ligand is sensitive, switch from a covalent amine coupling to a capture-based method (e.g., His-tag capture) that is easier to regenerate [58].
BLI Troubleshooting Guide
Frequently Asked Questions
Q1: The baseline is unstable or shows a lot of noise. This is a common issue in BLI and can be caused by:
- Sensor hydration: Ensure biosensors are properly hydrated according to the manufacturer's protocol before use [61].
- Plate seal integrity: In long experiments, sample evaporation in microplate wells can cause signal drift. Use proper plate seals to prevent evaporation [59].
- Sensor compatibility: Verify that the chosen biosensor is compatible with your sample buffer. Viscous solutions or high detergent concentrations can sometimes cause instability [13].
Q2: The binding signal is weak, even with a high analyte concentration. A weak signal can result from:
- Inefficient ligand loading: The ligand may not be immobilized at a high density or in the correct orientation. Try different loading conditions or a different biosensor type [61] [60].
- Low molecular weight analyte: BLI sensitivity decreases for small molecules. For analytes <10 kDa, use specialized biosensors (e.g., Super-Streptavidin) and ensure a high ligand density to amplify the signal [61].
- Improper referencing: Always subtract the signal from a reference sensor (immobilized with a non-interacting molecule) to account for buffer effects and non-specific binding [60].
Q3: The calculated binding affinity does not match my other data (e.g., SPR, ITC). Differences in affinity between techniques are common due to technical artifacts:
- Mass transport limitation: If the association rate is very fast, the observed binding may be limited by how quickly the analyte diffuses to the sensor surface. Increase the shaking speed to enhance mixing [60].
- Avidity effects: Multivalent immobilization of the ligand can lead to avidity, making the interaction appear stronger. Use a lower ligand density or a monovalent capture system [61].
- Confirm with an orthogonal method: Use BLI for rapid screening, but validate key results with a complementary technique like SPR or ITC [59] [63].
Table 2: Summary of Common Issues and Solutions
| Problem | Potential Causes | Recommended Solutions |
|---|---|---|
| High Non-Specific Binding | - Inadequate surface blocking- Unsuitable buffer conditions | - Include BSA or casein in buffer [63]- Optimize salt/detergent concentration [63] |
| Poor Reproducibility | - Sample aggregation- Inconsistent immobilization | - Centrifuge/filter samples before run [13]- Standardize ligand coupling protocol [58] |
| Weak or No Signal | - Low ligand activity- Low molecular weight analyte | - Check ligand functionality- Use high-density sensors for small molecules [61] |
| Unreliable Kinetics | - Mass transport limitation- Sensor surface heterogeneity | - Increase flow rate (SPR) or shaking (BLI) [60]- Lower ligand density [61] |
Key Experimental Protocols
Protocol 1: Determining Binding Kinetics of an Antibody to a Membrane Protein Receptor Using SPR
This protocol outlines steps to characterize the interaction between a soluble antibody and a membrane protein (e.g., a GPCR) reconstituted in nanodiscs.
- Surface Preparation: Dock a CM5 sensor chip. Activate the carboxymethylated dextran surface with a 1:1 mixture of 0.4 M EDC and 0.1 M NHS for 7 minutes.
- Ligand Immobilization: Dilute the capture antibody (e.g., Anti-His) in 10 mM sodium acetate buffer (pH 4.5) and inject over the activated surface for a target immobilization of 5000-8000 RU. Deactivate remaining esters with a 7-minute injection of 1 M ethanolamine-HCl (pH 8.5).
- Receptor Capture: Inject the His-tagged membrane protein in nanodiscs over the capture surface for 2-3 minutes to achieve a consistent capture level of ~100 RU.
- Kinetic Measurement:
- Baseline: Stabilize with HBS-EP+ running buffer for 2-3 minutes.
- Association: Inject a 3-fold dilution series of the antibody (e.g., from 100 nM to 1.2 nM) for 5 minutes.
- Dissociation: Switch to running buffer for 10-15 minutes.
- Regeneration: Remove the captured receptor complex with a 30-second pulse of 10 mM glycine (pH 1.5). The capture surface is ready for a new cycle.
- Data Analysis: Double-reference the sensorgrams (reference surface and buffer injections). Fit the data to a 1:1 binding model to extract (k{on}), (k{off}), and (K_D).
Protocol 2: Screening Small Molecule Inhibitors Against a Viral Protein Using BLI
This protocol describes a high-throughput method to screen for compounds that disrupt a protein-protein interaction.
- Biosensor and Plate Preparation: Hydrate Anti-His (HIS1K) biosensors in a black 96-well plate containing kinetic buffer (KB) for at least 10 minutes.
- Ligand Loading: Dispense a solution of the His-tagged viral protein (10 µg/mL in KB) into a microplate. Perform the "Load" step for 300 seconds to immobilize the protein.
- Baseline Equilibration: Transfer the biosensors to a column containing only KB for 60 seconds to establish a stable baseline.
- Association of Binding Partner:
- Control Step: Dip biosensors into a well with the known protein binding partner to record a positive control signal.
- Dissociation: Move biosensors to KB for 5 minutes to measure dissociation.
- Inhibitor Screening:
- Regeneration: Strip the biosensors with 10 mM glycine (pH 2.0) and re-load with fresh viral protein.
- Competition Step: Pre-incubate the protein binding partner with individual small molecules from a compound library. Dip the loaded biosensors into these mixtures.
- A reduction in the binding signal compared to the control (Step 4) indicates inhibition.
- Data Analysis: Use the system software to quantify the binding response in the competition step. Hits are identified as compounds that reduce the binding signal by >50%.
Essential Research Reagent Solutions
The following table lists key materials required for successful SPR and BLI experiments.
Table 3: Key Research Reagents and Their Functions
| Reagent / Material | Function | Example Product Codes |
|---|---|---|
| SPR Sensor Chips | Provides the gold surface with various chemistries for ligand immobilization. | CM5 (carboxymethylated dextran), NTA (nitrilotriacetic acid), L1 (lipophilic) |
| BLI Biosensors | Disposable fiber-optic tips with surface chemistry for ligand capture. | Streptavidin (SA), Anti-Human IgG Fc (AHQ), Ni-NTA (NTA), Anti-His (HIS1K) [61] |
| EDC / NHS | Cross-linking reagents for covalent amine coupling on carboxymethylated surfaces. | Common SPR coupling kit components |
| Running Buffer (HBS-EP+) | Standard buffer (HEPES, Saline, EDTA, Polysorbate 20) for maintaining sample and surface stability. | Cytiva BR100669 |
| Regeneration Solutions | Buffers (e.g., low pH, high salt) to remove bound analyte without damaging the immobilized ligand. | 10 mM Glycine-HCl (pH 1.5-3.0), 1-4 M MgCl₂ |
| Black Microplates | Low-evaporation, optical-bottom plates for BLI assays to minimize background signal. | Greiner Bio-One 655209, Sartorius 18-5166 [61] |
From Pitfalls to Performance: An Optimization Guide for Robust Results
For researchers studying membrane protein interactions, the initial step of cell lysis is a critical juncture. The choice of lysis buffer dictates a fundamental trade-off: efficient solubilization of the target protein versus preservation of its native structure and interactions. Stringent denaturing conditions can efficiently extract challenging membrane proteins but often at the cost of disrupting the very protein complexes that are the subject of study. This technical support center provides troubleshooting guides and FAQs to help you navigate this balance, directly supporting advanced research in drug development and membrane protein biology.
FAQ: Core Principles of Lysis Buffer Selection
Q1: Why is lysis buffer choice so critical for studying protein complexes?
The lysis buffer is the first chemical environment your protein complexes encounter outside the intact cell. Its composition directly determines whether these complexes remain intact for analysis. While both mild and strong buffers are suitable for generating whole-cell extracts for Western blotting, only milder, non-denaturing buffers are generally recommended for co-immunoprecipitation (co-IP) experiments where protein-protein interactions must be preserved [64].
Q2: What is the fundamental difference between RIPA and NP-40 buffers?
The key difference lies in their stringency, or their ability to denature proteins and disrupt interactions. RIPA buffer is a stronger, partially denaturing buffer because it contains the ionic detergent Sodium Deoxycholate, which helps disrupt nuclear membranes and solubilize cellular components more aggressively [64]. In contrast, NP-40 buffer is a milder, non-ionic detergent that effectively solubilizes the cell membrane but is less likely to disrupt protein-protein interactions, making it preferable for native complex purification [65] [66].
Q3: How does buffer selection differ for cytoplasmic versus membrane-bound proteins?
Proteins located in different cellular compartments often require different lysis conditions for optimal extraction. The table below provides a general guide for matching the lysis buffer to the protein's cellular location.
Table: Lysis Buffer Selection Based on Protein Localization
| Protein Location | Recommended Lysis Buffer |
|---|---|
| Whole Cell | NP-40, RIPA [66] |
| Cytoplasmic | Cytoplasmic and Nuclear Protein Extraction Kit [66] |
| Membrane-bound | NP-40, RIPA [66] |
| Nuclear | Cytoplasmic and Nuclear Protein Extraction Kit [66] |
| Mitochondria | RIPA [66] |
| Golgi Apparatus | Enhanced RIPA [66] |
Q4: What are the essential additives in a lysis buffer and what are their functions?
A well-formulated lysis buffer contains several key components that work together to extract and stabilize proteins.
Table: Key Components of a Lysis Buffer and Their Functions
| Component | Function |
|---|---|
| Detergents | Break down cell membranes and solubilize proteins; type determines stringency [66]. |
| Buffers | Provide a stable pH environment to maintain protein structure and function [66]. |
| Salts | Maintain physiological ionic strength and stabilize protein structure [66]. |
| Protease Inhibitors | Prevent proteins from being degraded by proteases during and after lysis [66]. |
| Phosphatase Inhibitors | Protect the phosphorylation state of proteins, crucial for signaling studies [64]. |
| Chelating Agents | Bind metal ions, inhibiting metal-dependent proteases [66]. |
Troubleshooting Guide: Common Lysis-Related Problems
Table: Troubleshooting Common Lysis Problems in Protein Interaction Studies
| Problem | Possible Causes | Recommendations |
|---|---|---|
| Low/No Signal in Co-IP | 1. Protein Interactions Disrupted: Lysis buffer is too stringent (e.g., using RIPA for co-IP) [64].2. Low Protein Expression: Target protein is expressed at levels below detection [64]. | 1. Switch to a milder lysis buffer like Cell Lysis Buffer #9803. Ensure sonication is performed to shear DNA and aid extraction [64].2. Check protein expression levels in input lysate controls. Use expression profiling tools to confirm expression in your model system [64]. |
| Low Yield of Target Protein | 1. Incomplete Lysis: Insufficient detergent strength or mechanical disruption [67].2. Protein Insolubility: Target protein may form inclusion bodies (in bacterial expression) [67]. | 1. Optimize lysis protocol; combine chemical and physical methods. For viscous lysates, add nucleases to digest DNA [67].2. Optimize expression conditions (e.g., lower temperature). For inclusion bodies, use denaturation and refolding strategies [67]. |
| Non-specific Binding | Off-target proteins binding non-specifically to beads or the antibody itself [64]. | Include a bead-only control and an isotype control to identify the source of background. Pre-clear the lysate if necessary [64]. |
| Protein Degradation | Proteases released during lysis are degrading the target protein [68]. | Always work on ice and use pre-chilled buffers. Include a broad-spectrum protease inhibitor cocktail in your lysis buffer [66] [68]. |
Key Experimental Protocols
Protocol 1: Lysis of Adherent Mammalian Cells for Co-IP
This protocol is designed for the gentle extraction of proteins while preserving interactions.
- Harvest & Wash: Culture and treat cells as required. Remove culture medium and wash cells with ice-cold Phosphate-Buffered Saline (PBS). Remove PBS completely [66].
- Lyse: Add an appropriate volume of chilled, additive-supplemented mild lysis buffer (e.g., based on NP-40 or a specialized cell lysis buffer) directly to the culture vessel. Use approximately 200-400 µL per well of a 6-well plate. Incubate on ice for 30 minutes with occasional gentle agitation [66] [64].
- Sonicate (Crucial for Co-IP): Briefly sonicate the lysates on ice. This ensures nuclear rupture, shears genomic DNA (reducing viscosity), and improves protein recovery, particularly for nuclear and membrane proteins, without disrupting most protein complexes [64].
- Clarify: Transfer the lysate to a microcentrifuge tube. Centrifuge at 14,000 x g for 10 minutes at 4°C to pellet insoluble debris [66].
- Collect: Carefully transfer the supernatant (the soluble protein extract) to a new tube. Proceed immediately to immunoprecipitation or store at -80°C.
Protocol 2: Sequential Denaturation for Ligand Target Identification
For applications like drug target screening where ligand-induced stability is measured, sequential denaturation can greatly enhance sensitivity compared to one-step methods [69].
- Sample Preparation: Lyse cell pellets in an ice-cold PBS-based lysis buffer containing protease inhibitors. Clarify the lysate by centrifugation [69].
- Ligand Incubation: Incubate aliquots of the lysate with either the ligand of interest (e.g., a drug) or a vehicle control.
- Sequential Denaturation: Subject the same individual sample to multiple, sequential denaturation treatments. For example, the SDPP (TEMP-SL) method involves:
- Centrifugation & Analysis: After each denaturation step, centrifuge the sample to separate the precipitated proteins from the soluble ones. Analyze the soluble fractions by mass spectrometry to identify proteins whose solubility was preserved by ligand binding across multiple denaturation conditions [69]. This cumulative effect amplifies the detection signal for target identification.
The Scientist's Toolkit: Research Reagent Solutions
Table: Essential Reagents for Protein Extraction and Analysis
| Reagent / Kit | Primary Function | Key Applications |
|---|---|---|
| NP-40 Lysis Buffer | Mild, non-ionic detergent-based lysis; preserves protein-protein interactions [65] [66]. | Co-IP, extraction of cytoplasmic and membrane proteins, native complex studies. |
| RIPA Lysis Buffer | Strong, partially denaturing lysis; contains ionic detergents for efficient membrane disruption [65] [66]. | Total protein extraction, challenging membrane proteins, Western blotting (non-interaction studies). |
| Cytoplasmic/Nuclear Extraction Kit | Sequential lysis for fractionation of cellular components. | Studying protein localization, transcription factors, signaling in specific compartments. |
| Protease Inhibitor Cocktail | Broad-spectrum inhibition of serine, cysteine, metallo-, and acid proteases. | All protein extraction procedures to prevent degradation. |
| Phosphatase Inhibitor Cocktail | Inhibition of serine/threonine and tyrosine phosphatases. | Phosphoprotein studies, signal transduction pathway analysis. |
| Micrococcal Nuclease / DNase I | Digestion of DNA in lysates to reduce viscosity. | All lysis protocols, especially for nuclear-rich samples, to improve handling [67]. |
Workflow and Pathway Visualizations
Lysis Buffer Selection Workflow
The following diagram outlines a logical decision pathway for selecting the appropriate lysis strategy based on experimental goals.
Protein Solubility Shift Assay
This diagram illustrates the conceptual workflow of stability shift-based assays like SDPP or CETSA, used for identifying drug targets.
Core Principles of IgG-Binding Proteins
Q: What are the fundamental differences between Protein A and Protein G?
Protein A and Protein G are bacterial proteins that bind to the Fc region of antibodies, primarily IgG, but they differ in their origin, structure, and binding specificity [70] [71]. These differences are critical for selecting the right tool for antibody purification or immunoprecipitation.
The key distinction lies in their binding profiles across species and IgG subclasses [70]. Protein G generally exhibits a broader binding capacity for mouse and human IgG subclasses compared to Protein A. Specifically, Protein G is superior for binding mouse IgG1 and human IgG3, which bind weakly or not at all to Protein A [70] [72].
Q: How does the choice of bead impact my experiment in membrane protein research?
In the context of membrane protein research, where target abundance is often low and sample preparation can introduce impurities, the choice of bead is crucial for success. Selecting a bead with low affinity for your target antibody can lead to:
- Poor Yield: Inefficient capture of the antibody-antigen complex.
- High Background: Non-specific binding, obscuring genuine protein interactions.
- Failed Experiments: Inability to reliably detect membrane protein interactions.
Using the optimal bead ensures efficient immunoprecipitation, maximizing the signal-to-noise ratio for downstream detection methods like Western blot, which is particularly important when studying low-abundance membrane protein complexes [73] [74].
Comparative Binding Data
Q: Where can I find a detailed comparison of binding affinities?
The tables below summarize the binding specificities of Protein A and Protein G for various species and IgG subclasses, providing a guide for selection. A "+" symbol indicates binding strength.
| Species | Immunoglobulin | Protein A Binding | Protein G Binding |
|---|---|---|---|
| Human | IgG (normal) | ++++ | ++++ |
| IgG1 | ++++ | ++++ | |
| IgG2 | ++++ | ++++ | |
| IgG3 | - | ++++ | |
| IgG4 | ++++ | ++++ | |
| Mouse | IgG1 | + | ++++ |
| IgG2a | ++++ | ++++ | |
| IgG2b | +++ | +++ | |
| IgG3 | ++ | +++ |
| Species | Immunoglobulin | Protein A Binding | Protein G Binding |
|---|---|---|---|
| Rabbit | IgG | ++++ | +++ |
| Cow | IgG | +/- | ++ |
| Goat | IgG | +/- | ++ |
| Sheep | IgG | +/- | ++ |
| Rat | IgG1 | - | + |
| IgG2a | - | ++++ | |
| IgG2b | - | ++ | |
| Pig, Dog, Cat | IgG | Recommended | Not Recommended |
Troubleshooting Guide
Q: My immunoprecipitation yield is low. Could the bead choice be the issue?
Yes. Low yield is often directly linked to a mismatch between the bead and your antibody. Please consult the workflow below to troubleshoot your experiment.
Q: I am getting high non-specific binding. What steps can I take?
High background can be mitigated by optimizing your protocol.
- Pre-clear Your Lysate: Incubate the sample with bare beads or control beads to remove proteins that bind non-specifically.
- Optimize Wash Stringency: Increase the salt concentration (e.g., 150-500 mM NaCl) or add mild detergents (e.g., 0.1% Triton X-100) to your wash buffers.
- Verify Bead Quality: Ensure beads are fresh and properly stored. Bead degradation can increase non-specific binding.
Experimental Protocols
Q: What is a standard protocol for immunoprecipitation using these beads?
The following workflow provides a general framework for an immunoprecipitation experiment. Always optimize conditions for your specific protein and antibody pair.
Detailed Methodology:
- Prepare Cell Lysate: Lyse cells in a suitable lysis buffer (e.g., RIPA buffer) supplemented with protease inhibitors. Keep samples on ice. Clarify the lysate by centrifugation at >12,000 x g for 15 minutes at 4°C to remove insoluble debris [74].
- Bind Antibody to Beads: Wash the recommended amount of Protein A or G beads with lysis buffer. Incubate the beads with your specific primary antibody for 1-2 hours at 4°C with gentle mixing. A typical ratio is 1-10 µg antibody per 10-50 µl of bead slurry.
- Incubate Lysate with Beads: Add the pre-cleared lysate to the antibody-bound beads. Incubate for 2-4 hours or overnight at 4°C with gentle mixing to form the immune complex.
- Wash Beads: Pellet the beads by brief centrifugation and carefully remove the supernatant. Wash the beads 3-4 times with 1 ml of ice-cold lysis buffer to remove unbound and non-specifically bound proteins.
- Elute Target Protein: After the final wash, elute the bound target protein by boiling the beads in 2X Laemmli SDS-PAGE sample buffer for 5-10 minutes. The eluate can then be analyzed by Western blot [73] [74].
Frequently Asked Questions (FAQs)
Q: When should I consider using Protein A/G? Protein A/G is a recombinant fusion protein that combines the IgG binding domains of both Protein A and Protein G [70] [75]. It is ideal when you work with a variety of antibodies from different species or when the specific subclass of an antibody is unknown, as it binds a broader range of IgG subclasses than either Protein A or G alone [70].
Q: What about Protein L? Protein L binds to antibodies through the kappa light chain, not the Fc region [70] [71]. This allows it to bind a wider range of immunoglobulin classes (IgG, IgM, IgA, IgE, and IgD) but only if they contain kappa light chains [70]. It is particularly useful for purifying or capturing recombinant antibody fragments like scFvs or Fab fragments that lack an Fc region.
Q: How do I handle and store these beads? Resuspend the beads by gentle vortexing or inversion. For long-term storage, keep at 4°C. Do not freeze. Beads should be stored in a preservative solution provided by the manufacturer and should not be allowed to dry out.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents for Antibody-Based Applications
| Item | Function | Key Considerations |
|---|---|---|
| Protein A Beads | Affinity purification of antibodies, IP. | Ideal for rabbit, human (except IgG3), mouse (IgG2a, 2b, 3), pig, dog, and cat IgG [70] [71]. |
| Protein G Beads | Affinity purification of antibodies, IP. | Superior for human (all subclasses), mouse (all subclasses, esp. IgG1), rat, cow, goat, and sheep IgG [70] [72]. |
| Protein A/G Beads | Broad-spectrum antibody purification and IP. | Recommended when antibody specificity is unknown or for labs handling a wide variety of antibodies [70] [75]. |
| Protein L Beads | Binding to kappa light chains of antibodies. | Use for antibody fragments (Fabs, scFvs) or full-length Igs (IgA, IgM, IgD) that do not bind Protein A/G [70] [71]. |
| Crosslinked Beaded Agarose | A common solid support for column-based affinity purification of antibodies [70]. | Provides a stable, porous matrix for high-capacity binding. |
| Magnetic Beads | A popular support for immunoprecipitation applications, enabling easy separation using a magnet [70]. | Ideal for high-throughput applications and rapid protocol steps. |
Epitope masking and IgG signal interference present significant challenges in biomedical research, particularly in the fields of immunology, drug development, and diagnostics. These phenomena can obscure accurate detection of target proteins, compromise assay sensitivity, and lead to false negative results. This technical support center provides targeted troubleshooting guides and FAQs to help researchers identify, understand, and overcome these limitations within the broader context of advancing membrane protein interaction detection research.
FAQs and Troubleshooting Guides
FAQ 1: What is epitope masking and how does it interfere with immunoassays?
Answer: Epitope masking occurs when existing antibodies or other molecules bind to and physically block the specific regions (epitopes) on an antigen that other antibodies need to access for detection. This creates interference in immunoassays by:
- Steric Hindrance: Antibodies, particularly intact IgM with its large pentameric structure, can effectively compete with detection antibodies for binding sites due to their high avidity and ability for polyvalent binding to repetitive epitopes like those on viral capsids [76].
- Assay Signal Suppression: Co-existing antibodies can cause significant interference in immune complex (IC) assay formats, leading to suppressed detection signals and reduced assay sensitivity, particularly at early time points in immune response monitoring [76].
- B Cell Response Modulation: In biological systems, epitope masking by existing antibodies can negatively feedback on B cell responses to masked epitopes while potentially diversifying responses to unmasked, often subdominant, epitopes [77] [78].
FAQ 2: Why does my immune complex assay show unexpectedly low IgG signals despite confirmed antibody presence?
Answer: This common issue typically stems from IgM interference, especially in samples collected during early immune responses when IgM titers are high. The underlying mechanism involves:
- Competitive Binding: Polymeric IgM antibodies achieve high avidity despite low binding affinity, making them particularly effective at competing with IgG for epitope binding, especially on antigens with repetitive epitope structures [76].
- Time-Dependent Effect: The interference is most prominent in samples collected within the first 4 weeks after antigen exposure when IgM responses peak, while samples from later timepoints typically show minimal interference [76].
Solution: Implement selective IgM digestion prior to IgG detection:
- Treat serum samples (10 μL) with IgM-cleaving protease (1 μL, final concentration 10 U/μL) overnight at room temperature [76].
- Proceed with your standard IgG immune complex assay protocol.
- This approach has been shown to increase anti-AAV2 and anti-AAV9 IgG signals when corresponding IgM antibodies are present in the matrix [76].
FAQ 3: How can I prevent epitope masking in membrane protein-ligand interaction studies?
Answer: Membrane proteins present particular challenges due to their structural complexity and hydrophobic nature. Advanced strategies integrating mass spectrometry with membrane mimetics offer solutions:
- Membrane Mimetic Systems: Utilize nanodiscs, peptidiscs, or styrene-maleic acid (SMA) polymers to create more native-like environments that maintain functional protein conformations and ligand-binding sites while reducing non-specific masking [79].
- MS Methodology Innovations: Implement native mass spectrometry, nativeomics, solution-phase thermochemistry, or ion mobility-mass spectrometry (IM-MS) to better preserve intact assemblies and capture co-bound lipids and ligands [79].
- Detergent Optimization: Employ tailored detergent architectures and MS-compatible detergents to improve solubilization while maintaining epitope accessibility [79].
FAQ 4: What experimental strategies can overcome epitope masking in therapeutic antibody development?
Answer: Several engineering approaches can circumvent masking issues:
- Conditional Activation: Engineer masked antibodies with protease-cleavable domains that remain inactive in healthy tissues but become activated in disease microenvironments (e.g., by MMP-9 in tumors) [80].
- Multi-Specific Targeting: Develop bispecific antibodies that engage multiple epitopes or antigens simultaneously, reducing the impact of masking on any single epitope [81].
- Epitope Diversification: Focus on subdominant epitopes that are less likely to be masked by pre-existing antibody responses, thereby broadening the protective response [82] [78].
Experimental Protocols
Protocol 1: IgM Protease Digestion for Improved IgG Detection
Purpose: To eliminate IgM interference in IgG-specific immune complex assays [76].
Materials:
- IgM-cleaving protease (e.g., IgMBRAZOR)
- Preclinical serum samples
- Phosphate buffered saline (PBS)
- Standard reagents for your immune complex assay
Procedure:
- Prepare serum samples (10 μL) in duplicate for protease treatment and untreated control.
- Experimental group: Add IgM-cleaving protease (1 μL, final concentration 10 U/μL) to serum samples.
- Control group: Add PBS (1 μL) to serum samples instead of protease.
- Incubate overnight at room temperature.
- Proceed with your standard IgG immune complex assay protocol:
- Incubate samples with capsid particles or target antigens
- Incubate with biotinylated capture antibody (0.5 µg/mL)
- Transfer to streptavidin-coated microtiter plate
- Detect captured complexes with HRP-labeled detection antibody
- Compare signals between protease-treated and untreated samples to assess IgM interference.
Protocol 2: Evaluation of Masking Efficiency in Engineered Antibodies
Purpose: To validate the efficiency of masking strategies in conditionally active therapeutic antibodies [80].
Materials:
- Masked antibody construct (e.g., CaM-CBP peptide clamp fused to antibody)
- Appropriate protease for demasking (e.g., MMP-9)
- Antigen-positive cells
- Flow cytometry equipment
- ADCC reporter assay system
Procedure:
- On-Cell Binding Assay:
- Incubate various concentrations of masked antibodies with antigen-positive cells
- Measure binding efficiency via flow cytometry
- Compare with unmodified antibodies to calculate fold-reduction in EC50
- Functional Masking Validation:
- Perform antibody-dependent cellular cytotoxicity (ADCC) assay with masked constructs
- Use effector cells (e.g., NK cells) and target cells expressing the antigen
- Measure effector cell activation and compare with unmodified antibodies
- Protease Activation Test:
- Treat masked antibodies with relevant protease (e.g., MMP-9)
- Repeat binding and functional assays to confirm restoration of activity
- Calculate fold-reduction in EC50 pre- and post-demasking
Table 1: Effectiveness of IgM Digestion in Improving IgG Detection in Immune Complex Assays
| Assay Target | Sample Type | IgM Digestion Impact on IgG Detection | Detection Rate Improvement |
|---|---|---|---|
| Anti-AAV2 IgG | Cynomolgus serum | Signal increase when anti-AAV2 IgM present | Day 15: 36% to 82% positive [76] |
| Anti-AAV9 IgG | Cynomolgus serum | Signal increase correlated with IgM strength | Day 7: 28% to 39% positive [76] |
Table 2: Performance Metrics of Antibody Masking Strategies
| Masking Approach | Application | Efficiency (Fold-Reduction in EC50) | Protease Activation |
|---|---|---|---|
| CaM-CBP Peptide Clamp | Trastuzumab/Cetuximab | Up to 410x (Binding) [80] | MMP-9 cleavable |
| CaM-CBP Peptide Clamp | Trastuzumab/Cetuximab | Up to 78x (ADCC function) [80] | MMP-9 cleavable |
| Coiled-coil Domains | HER2/CD19 targeting | 753x (Binding) [80] | MMP-2/9 cleavable |
Research Reagent Solutions
Table 3: Essential Reagents for Combating Epitope Masking and Interference
| Reagent | Function | Example Applications |
|---|---|---|
| IgM-cleaving protease | Selective digestion of IgM antibodies | Eliminating IgM interference in IgG IC assays [76] |
| Calmodulin-CBP masking system | Human-derived protein-based masking domain | Creating conditionally active therapeutic antibodies [80] |
| Matrix metalloproteinases (MMP-2/9) | Tumor microenvironment proteases | Activating masked antibodies in diseased tissue [80] |
| Membrane mimetics (Nanodiscs, SMA polymers) | Creating native-like membrane environments | Maintaining MP structure and ligand accessibility [79] |
| Bispecific antibody formats | Simultaneous targeting of multiple epitopes | Reducing escape mutations and epitope masking [81] |
Visualized Workflows and Pathways
IgM Interference Troubleshooting
Conditional Antibody Activation
Why Are Controls Critical in Co-Immunoprecipitation?
In co-immunoprecipitation (co-IP) and related techniques, controls are not merely optional; they are fundamental for validating your results. They are the cornerstone of experimental integrity, allowing you to distinguish specific biological signals from non-specific background and experimental artifacts [83] [84]. Without the proper controls, you cannot be confident that an observed protein-protein interaction is real. This is especially critical in membrane protein research, where interactions can be transient and detection is often challenged by background noise [85].
This guide details the three essential controls—Input Lysate, Bead-Only, and Isotype Control—to ensure the specificity and reliability of your co-IP experiments.
Input Lysate Control
What is it?
The input lysate control consists of a small portion (typically 1-10%) of the original cell or tissue lysate set aside before any immunoprecipitation steps are performed [10] [86]. This sample represents the total starting material.
Why is it essential?
The input control serves multiple critical functions as a positive control and a quality assessment tool [10] [87]:
- Verifies Protein Presence: It confirms that your protein of interest (the "bait") and any suspected interacting partners (the "prey") were present and detectable in your original sample.
- Confirms Negative Results: If a prey protein is detected in the input but is absent in the co-IP sample, it provides evidence that an interaction was not captured, rather than the prey being absent altogether [10].
- Assays Antibody Efficiency: By comparing the band intensity of the target protein in the IP lane to its band in the input lane, you can gauge how effectively your antibody precipitated the target [10].
- Prepare your cell or tissue lysate as usual [86].
- Before adding any antibodies or beads, remove 1-10% of the total lysate volume.
- Dilute this aliquot with an equal volume of 2X Laemmli buffer.
- Boil the input sample for 5-10 minutes, then store at -20°C until you are ready to run your western blot [86].
- Proceed with the immunoprecipitation on the remaining lysate.
- During western blot analysis, run the input sample alongside your IP samples.
The following workflow integrates the preparation and use of the input lysate control into a standard co-IP experiment:
Bead-Only Control
What is it?
The bead-only control is a sample where the lysate is incubated with plain, unconjugated beads (e.g., Protein A or G agarose/sepharose) without any antibody present [87].
Why is it essential?
This control identifies proteins that bind non-specifically to the beads themselves or to the bead matrix [87]. Beads can have inherent "stickiness" for certain proteins, and this control allows you to account for that background. If you see a band in your experimental co-IP that is also present in the bead-only control, it is likely a non-specific interaction and not a true binding partner.
- Take an equal volume of your pre-cleared lysate and add it to a fresh tube.
- Add the same amount and type of beads you will use in your experimental IP, but do not add any antibody.
- Incubate this mixture on a rotator at 4°C for the same duration as your experimental IP.
- Proceed with all the same washing and elution steps.
- Analyze the eluate alongside your experimental sample by western blot.
Isotype Control
What is it?
An isotype control is an antibody that matches the host species, immunoglobulin class, subclass, and conjugation (e.g., fluorophore, biotin) of your primary IP antibody but lacks specificity for your target protein [83] [84]. It is a negative control that should not immunoprecipitate your protein of interest.
Why is it essential?
The isotype control identifies non-specific binding caused by the antibody itself, particularly interactions mediated by the Fc region with cellular proteins, lipids, or Fc receptors [83] [84]. Any signal observed in the isotype control lane indicates background staining that is not due to the specific antigen-binding region of your primary antibody.
How to Choose the Right Isotype Control
Selecting a matched isotype control is critical for a meaningful result [83] [84]:
| Your Primary Antibody Characteristics | Your Required Isotype Control |
|---|---|
| Host Species (e.g., Mouse) | Mouse antibody |
| Isotype & Subclass (e.g., IgG1) | Mouse IgG1 antibody |
| Conjugation (e.g., Unconjugated) | Unconjugated Mouse IgG1 |
| Conjugation (e.g., FITC) | Mouse IgG1-FITC |
- Split your lysate into two equal aliquots.
- To one tube, add your specific primary antibody (experimental IP).
- To the other tube, add the isotype control antibody at the same concentration.
- Add the same type and amount of beads to both tubes.
- Process both samples identically through incubation, washing, and elution.
- Analyze both eluates by western blot. A true interacting partner will appear in the experimental IP lane but not in the isotype control lane.
The following decision diagram helps interpret results by incorporating all three essential controls:
Troubleshooting Guide: Interpreting Your Controls
This table outlines common problems identified by controls and their potential solutions.
| Problem Scenario | Possible Cause | Recommended Solution |
|---|---|---|
| Target protein is absent in Input Lysate | Low protein expression or inefficient lysis [87]. | Verify expression in your cell line/tissue. Optimize lysis buffer; use a milder, non-denaturing buffer for co-IP [87] [86]. Include protease inhibitors [86]. |
| Prey protein is in Input but absent in co-IP | Protein-protein interaction was disrupted or is transient [10]. | Use a milder lysis buffer (avoid RIPA) [87]. Try crosslinking. Verify the antibody does not block the interaction site [10]. |
| Bands present in Isotype Control | Non-specific binding to the antibody's Fc region [83] [84]. | Pre-clear lysate with beads. Include Fc receptor blocking reagents. Titrate antibody to optimal concentration [83]. |
| Bands present in Bead-Only Control | Non-specific binding to the beads [87]. | Pre-clear lysate. Change bead type or supplier. Increase stringency of washes (e.g., increase salt concentration). |
| High background across all controls | Inadequate washing or antibody concentration too high. | Increase number and/or volume of washes. Optimize antibody concentration through titration. |
The Scientist's Toolkit: Essential Research Reagent Solutions
| Reagent / Material | Function in Control Experiments |
|---|---|
| Protein A/G Beads | Solid support for immobilizing antibody-antigen complexes; used in Bead-Only control [87]. |
| Isotype Control Antibody | Negative control antibody matching host species, isotype, and conjugation of primary antibody [83] [84]. |
| Non-denaturing Lysis Buffer | Preserves native protein-protein interactions during cell lysis (e.g., NP-40 based buffers) [87] [86]. |
| Protease/Phosphatase Inhibitor Cocktail | Prevents degradation and dephosphorylation of proteins during lysate preparation [86]. |
| Laemmli Buffer | Denatures proteins for accurate analysis by SDS-PAGE and western blot; used to prepare the Input Lysate control [86]. |
By rigorously implementing these controls, you can significantly enhance the validity and impact of your research on membrane protein interactions.
The study of membrane proteins, which are primary targets for a majority of marketed therapeutics, is fraught with technical challenges. A significant bottleneck in this research is the inherent instability of these proteins once extracted from their native membrane environment. During cell lysis, the carefully controlled cellular compartmentalization is disturbed, releasing endogenous proteases and phosphatases that can rapidly degrade proteins and remove essential post-translational modifications (PTMs) such as phosphorylation [88] [13]. This degradation leads to reduced protein yield, loss of activity, and biologically meaningless representations of protein activation states, critically hampering interaction studies and functional assays [88] [89].
The preservation of PTMs is not merely about protein stability; it is fundamental to understanding signal transduction. Phosphorylation is one of the most common and dynamic PTMs, with phosphatases responsible for removing phosphate groups from serine, threonine, and tyrosine residues [88] [90]. For membrane proteins, which are key players in cellular communication, preserving this phosphorylation status is essential for accurate analysis of their function and interactions [13]. Therefore, the use of tailored protease and phosphatase inhibitor cocktails is not just a routine step but a crucial strategy to overcome the limitations in membrane protein interaction detection research, ensuring that the data generated reflect the true in vivo state of the protein.
Understanding the Threats: Proteases and Phosphatases
Proteases (Proteolytic Enzymes)
Proteases are enzymes that hydrolyze peptide bonds, breaking down proteins. They are ubiquitous in all living organisms and are normally tightly regulated within cells [88] [91]. They are broadly categorized based on their catalytic mechanism and their site of cleavage:
- Endopeptidases: Cleave peptide bonds at internal sites within the protein chain. These include:
- Serine proteases
- Cysteine proteases
- Aspartic acid proteases
- Metalloproteases
- Exopeptidases: Cleave amino acids from the N- or C-terminus of proteins (e.g., aminopeptidases, carboxypeptidases) [91].
Phosphatases
Phosphatases are hydrolase enzymes that remove phosphate groups from proteins and other molecules. They are vital regulators of signaling pathways, controlling processes like signal transduction, cell division, and apoptosis [88] [91]. They are classified based on their substrate specificity and optimal pH:
- Serine/Threonine Phosphatases: Target phospho-serine and phospho-threonine residues.
- Protein Tyrosine Phosphatases: Target phospho-tyrosine residues.
- Acid Phosphatases: Active in the acidic environment of lysosomes.
- Alkaline Phosphatases: Active in more basic cellular environments [91] [90].
The following diagram illustrates how these enzymes are released during cell lysis and degrade your protein sample, and how inhibitor cocktails protect against this threat.
The Scientist's Toolkit: Key Reagents and Their Functions
No single compound is effective against all classes of proteases and phosphatases. Therefore, a cocktail or mixture of several inhibitors is required for comprehensive protection [88]. The table below summarizes the most commonly used inhibitors, their targets, and working concentrations.
Table 1: Common Protease Inhibitors
| Inhibitor | Target Protease Class | Mechanism | Typical Working Concentration |
|---|---|---|---|
| AEBSF | Serine | Irreversible | 0.2 - 1.0 mM |
| Aprotinin | Serine | Reversible | 100 - 200 nM |
| E-64 | Cysteine | Irreversible | 1 - 20 µM |
| Leupeptin | Serine & Cysteine | Reversible | 10 - 100 µM |
| Pepstatin A | Aspartic Acid | Reversible | 1 - 20 µM |
| EDTA | Metalloproteases | Chelates Cations (Reversible) | 2 - 10 mM |
| Bestatin | Aminopeptidases | Reversible | 1 - 10 µM [88] [92] |
Table 2: Common Phosphatase Inhibitors
| Inhibitor | Target Phosphatase Class | Mechanism | Typical Working Concentration |
|---|---|---|---|
| Sodium Fluoride | Ser/Thr and Acidic Phosphatases | Irreversible | 1 - 20 mM |
| Sodium Orthovanadate | Tyrosine and Alkaline Phosphatases | Irreversible | 1 - 100 mM |
| β-Glycerophosphate | Ser/Thr Phosphatases | Reversible | 1 - 100 mM |
| Sodium Pyrophosphate | Ser/Thr Phosphatases | Irreversible | 1 - 100 mM [88] [92] |
Research Reagent Solutions
For practical application in the lab, researchers can utilize the following solutions:
- Ready-to-Use Inhibitor Cocktails: Commercial tablets or pre-mixed solutions that offer convenience and consistency. These are available as protease-only, phosphatase-only, or combined cocktails, with options that are EDTA-free (for metalloprotease-independent work) or mass spectrometry-compatible [91] [92].
- Individual Inhibitor Stocks: Researchers can prepare concentrated stock solutions of individual inhibitors (as listed in Tables 1 & 2) in appropriate solvents (e.g., water, methanol, DMSO) to create custom-tailored cocktails for specific experimental needs [88].
Experimental Protocol: Evaluating Inhibitor Cocktail Efficacy
The following methodology, adapted from published studies, allows researchers to quantitatively assess the effectiveness of their chosen inhibitor cocktail in protecting protein samples [91].
Aim: To validate the performance of a protease and phosphatase inhibitor cocktail in preventing protein degradation and dephosphorylation in a complex cell lysate.
Materials and Reagents:
- Cultured cells (e.g., HEK293, THP-1) or tissue of interest.
- Ice-cold Cell Lysis Buffer (e.g., RIPA, T-PER).
- Protease Inhibitor Cocktail (e.g., containing AEBSF, E-64, Leupeptin, Pepstatin A, EDTA).
- Phosphatase Inhibitor Cocktail (e.g., containing Sodium Fluoride, Sodium Orthovanadate, β-Glycerophosphate).
- BCA or Bradford Protein Assay Kit.
- Quenched-fluorescent protease substrates (e.g., for cysteine or serine proteases).
- Phosphatase activity assay kit or a universal phosphatase substrate.
- SDS-PAGE and Western Blot equipment.
- Antibodies for total and phosphorylated proteins (e.g., anti-ERK1/2 and anti-phospho-ERK1/2).
- Microplate reader capable of fluorescence detection.
Procedure:
Cell Lysis and Sample Preparation:
- Grow cells to 80-90% confluency and rinse with ice-cold PBS.
- Divide the cell pellet into three equal aliquots.
- Sample 1 (Positive Control): Lyse cells in lysis buffer without any inhibitors.
- Sample 2 (Test): Lyse cells in lysis buffer with the combined protease and phosphatase inhibitor cocktail.
- Sample 3 (Negative Control): Lyse cells in lysis buffer and immediately heat-inactivate at 95°C for 10 minutes to denature all enzymes.
- Incubate all lysates on ice for 10-30 minutes, then clarify by centrifugation at 10,000 x g for 10 minutes at 4°C. Collect the supernatants [91].
Protein Quantification and Normalization:
- Determine the protein concentration of each supernatant using a BCA or Bradford assay.
- Normalize all samples to the same protein concentration using lysis buffer.
Functional Activity Assays:
- Protease Inhibition Assay:
- In a black 96-well plate, mix equal volumes of each sample with a quenched-fluorescent protease substrate (e.g., for papain/cysteine proteases or trypsin/serine proteases).
- Incubate at 37°C for 1-2 hours.
- Measure fluorescence at the appropriate excitation/emission wavelengths (e.g., 380/460 nm for a cysteine protease substrate). High fluorescence indicates substrate cleavage and thus protease activity [91].
- Phosphatase Inhibition Assay:
- Incubate each sample with a phosphatase substrate (e.g., pNPP or a fluorescent substrate) in a suitable buffer.
- Incubate at 37°C for 1 hour.
- Measure the absorbance or fluorescence of the released product. High signal indicates phosphatase activity [91].
- Protease Inhibition Assay:
Analysis of Phosphoprotein Preservation (Western Blot):
- Mix normalized protein samples with SDS-PAGE loading buffer.
- Resolve proteins by SDS-PAGE and transfer to a PVDF membrane.
- Probe the membrane with antibodies against phosphorylated signaling proteins (e.g., phospho-AKT, phospho-ERK1/2) and then re-probe for total protein to confirm equal loading.
- Strong phospho-signal in the "Test" sample and a weak or absent signal in the "Positive Control" indicates effective preservation of phosphorylation by the inhibitors [91].
The workflow for this validation experiment is summarized in the diagram below.
Troubleshooting Guide & FAQs
This section addresses common problems researchers encounter when working with inhibitor cocktails, with a specific focus on membrane protein applications.
Frequently Asked Questions
Q1: Are protease inhibitors always necessary, or are phosphatase inhibitors only for phospho-work? Yes, protease inhibitors are nearly always required during cell lysis and protein extraction to prevent general protein degradation. Phosphatase inhibitors are specifically required when the phosphorylation state (activation state) of proteins is being investigated [88].
Q2: Why should I use a cocktail instead of a single inhibitor like PMSF? No single chemical is effective against all types of proteases and phosphatases. Proteases belong to several distinct evolutionary families (serine, cysteine, aspartic, metallo-), and a single inhibitor like PMSF only targets serine proteases. Using a cocktail ensures broad-spectrum protection against the diverse range of enzymes released upon lysis [88].
Q3: I am working with metalloproteins. What inhibitor should I avoid? You should use an EDTA-free inhibitor cocktail. EDTA is a chelating agent that targets metalloproteases by removing essential metal ions. If your protein of interest requires metal ions for stability or function, EDTA could inactivate it [92].
Troubleshooting Common Problems
| Problem | Possible Cause | Solution |
|---|---|---|
| Poor Phosphoprotein Detection | Phosphatase activity not fully inhibited; inhibitors degraded. | Use a fresh, broad-spectrum phosphatase cocktail. Confirm sodium orthovanadate has been properly activated (heated to pH 10 until colorless). Avoid repeated freeze-thaw cycles of inhibitor stocks [88] [90]. |
| High Background Proteolysis | Protease inhibitor cocktail is not broad-spectrum; incorrect concentration used. | Use a cocktail containing inhibitors for serine, cysteine, aspartic, and metalloproteases (e.g., AEBSF, E-64, Pepstatin A, EDTA). Verify that the final working concentration in your lysate is correct [88] [92]. |
| Loss of Membrane Protein Activity | Inhibitors interfering with protein function; detergent incompatibility. | For sensitive functional assays, titrate the inhibitor cocktail to the minimum effective concentration. Consider using purified, recombinant inhibitors like aprotinin instead of crude mixtures. Ensure detergent in lysis buffer is compatible with activity [89] [13]. |
| Inconsistent Results Between Preparations | Improper storage or handling of inhibitors; incomplete solubilization. | Always store stock solutions and cocktails as recommended by the manufacturer (often -20°C or below). Ensure tablets are fully dissolved and liquid cocktails are thoroughly mixed into the lysis buffer [91]. |
Within the challenging field of membrane protein research, where the integrity of the protein sample is paramount, the strategic use of protease and phosphatase inhibitor cocktails is a non-negotiable practice. These reagents are fundamental for overcoming the critical limitation of protein degradation and dephosphorylation that occurs post-lysis. By faithfully preserving the native state and post-translational modifications of membrane proteins, robust and biologically relevant data from interaction studies and functional assays can be achieved. As the toolkit for membrane protein characterization advances with techniques like mass photometry [13], the foundational step of sample protection with effective inhibitor cocktails will remain a cornerstone of successful research and drug development.
What is the primary function of pre-clearing a lysate, and when is it recommended?
Pre-clearing is an optional step in immunoprecipitation (IP) and co-immunoprecipitation (Co-IP) workflows designed to reduce non-specific binding and background signal [86] [93]. The process involves incubating the cell or tissue lysate with only the beads (e.g., protein A/G agarose or magnetic beads) or with beads coupled to a non-specific control antibody before performing the actual IP with your target-specific antibody [86] [93]. This step helps remove proteins and other factors in the lysate that bind nonspecifically to the beads or to antibodies in general.
While it can increase the purity of the isolated proteins, some protocols note that pre-clearing may not be necessary for Western blot detection unless a contaminating protein interferes with visualizing your protein of interest [86]. The need for pre-clearing can also depend on the solid support used; magnetic bead-based IP protocols often state that pre-clearing is usually not necessary due to the lower nonspecific binding characteristics of the beads [93].
What are the established controls for validating antibody specificity and how do they help?
Using appropriate controls is fundamental to distinguishing specific signal from false positives. The table below summarizes the core types of controls used in antibody-based experiments [94].
Table: Established Controls for Antibody-Based Experiments
| Control Type | Description | Primary Function |
|---|---|---|
| Positive Control Lysate | Lysate from a cell line or tissue sample known to express the target protein. | Verifies the staining protocol works and provides the expected sensitivity/specificity; confirms that negative results in test samples are accurate [94]. |
| Negative Control Lysate | Lysate from a cell line or tissue sample known not to express the target protein (e.g., knockout/knockdown samples). | Checks for non-specific antibody binding (false-positive results) [94]. |
| Isotype Control | An antibody of the same class (e.g., IgG) but with no specificity for the target protein. | Serves as a negative control for the IP itself, helping to distinguish specific from non-specific binding in co-IPs [86]. |
How can validating with multiple antibodies prevent false positives?
Relying on a single antibody can lead to misinterpretation due to off-target binding. Using multiple antibodies that recognize different epitopes on the same target protein or different subunits of a protein complex provides orthogonal validation.
This principle is powerfully illustrated in multiplex bead-based assays, such as those developed for SARS-CoV-2 antibody detection. These assays simultaneously measure the immune response against multiple distinct viral proteins (e.g., Nucleocapsid (NC), Spike (S), Receptor Binding Domain (RBD)) [95] [96]. A true positive is indicated by a coherent signal across multiple antigens, whereas a signal against only one antigen may indicate cross-reactivity or nonspecific binding, effectively flagging a potential false positive [95] [96]. This multi-parameter approach provides a much higher level of confidence in the results.
What are the best practices for lysate preparation to minimize false results?
Proper lysate preparation is the foundation for a successful IP and is critical for preserving native protein interactions while minimizing artifacts [86] [93].
- Use Appropriate Lysis Buffers: Choose a buffer that effectively solubilizes your target protein while maintaining protein-protein interactions. Non-ionic detergents like NP-40 and Triton X-100 are commonly used for gentle, non-denaturing lysis [86].
- Include Protease and Phosphatase Inhibitors: Always add protease inhibitor cocktails to prevent protein degradation, which can generate fragments and cause unexpected bands on a Western blot. If studying phosphorylated proteins, include phosphatase inhibitors [86].
- Keep Samples Cold: Perform all steps on ice or at 4°C to slow enzymatic activity and maintain complex stability [86].
- Determine Protein Concentration: Use an assay (e.g., Bradford or BCA) to quantify total protein, ensuring equal loading across experiments [86].
Table: Common Lysis Buffer Compositions
| Buffer Type | Typical Composition | Recommended Use |
|---|---|---|
| Mild Lysis Buffer (e.g., NP-40) | 150 mM NaCl, 1% NP-40, 50 mM Tris-HCl (pH 8.0) | Ideal for extracting cytoplasmic and membrane proteins while preserving protein-protein interactions [86]. |
| Harsh Lysis Buffer (e.g., RIPA) | 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS | Effective for disrupting nuclear proteins or breaking apart stubborn protein complexes; may disrupt some weak interactions [86]. |
Experimental Workflow for False Positive Mitigation
The following diagram illustrates a robust experimental workflow that integrates preclearing, controlled immunoprecipitation, and multi-antibody validation to ensure reliable results.
Research Reagent Solutions
Table: Essential Materials for Robust Co-IP and False Positive Mitigation
| Reagent / Tool | Function | Considerations |
|---|---|---|
| Magnetic Beads | Solid support for antibody immobilization and antigen capture. | Preferred for IP; offer ease of use, high reproducibility, purity, and are amenable to automation [93]. |
| Agarose Resin | Porous, beaded support for affinity purification. | Traditional IP support; high binding capacity but requires centrifugation and longer processing times [93]. |
| Protease Inhibitor Cocktail | Prevents proteolytic degradation of target proteins and complexes. | Essential for maintaining protein integrity during lysis and IP; should be added fresh to lysis buffer [86]. |
| Phosphatase Inhibitor Cocktail | Prevents dephosphorylation of proteins. | Critical for studying phosphoproteins or phosphorylation-dependent interactions [86]. |
| Isotype Control Antibody | Matched immunoglobulin from the same species with no target specificity. | Serves as the critical negative control for the IP to identify proteins that bind nonspecifically to the antibody or beads [86]. |
| Positive Control Lysate | Sample with a verified, high level of the target protein. | Validates that the entire IP and detection protocol is functioning correctly [94]. |
| Negative Control Lysate | Sample from a knockout or knockdown cell line lacking the target. | Confirms the specificity of the primary antibody by checking for absence of signal [94]. |
Beyond a Single Method: A Framework for Cross-Validation and Data Integration
Membrane proteins (MPs) are critical players in cellular processes such as signal transduction, immune response, and material transport. Consequently, they represent a major class of drug targets. However, their inherent hydrophobicity and complex association with lipid bilayers pose significant challenges for studying their interactions. This technical support guide is framed within a broader thesis aimed at addressing the key limitations in MP interaction detection research: balancing throughput (the number of interactions that can be tested in a given time) with biological relevance (the ability to capture interactions in a native or near-native in vivo environment). The following FAQs, comparative tables, and detailed protocols are designed to help you select and troubleshoot the most appropriate method for your research goals.
Frequently Asked Questions (FAQs)
FAQ 1: What are the primary considerations when choosing a technique to study membrane protein interactions? Your choice should be guided by three main factors:
- Goal: Are you screening for novel interaction partners or validating a specific suspected interaction?
- Throughput Needs: Do you require a high-throughput method to test hundreds or thousands of potential partners, or a lower-throughput method for detailed study?
- Biological Context: Is it essential to detect the interaction in a living cell (in vivo), or is a controlled in vitro environment sufficient? The need to preserve the native membrane environment is often a decisive factor.
FAQ 2: Why is the in vivo environment particularly important for studying membrane protein interactions? Membrane proteins are embedded in a complex lipid environment and their structure, function, and interactions can be heavily influenced by surrounding lipids and other cellular factors. Techniques that remove them from this context, using harsh detergents for example, can disrupt native conformations and lead to the loss of transient or weak interactions [97] [98]. In vivo and membrane-mimetic techniques help preserve these critical conditions.
FAQ 3: A suspected interaction partner for my membrane protein bait did not show up in my MYTH assay. What could be the cause? This is a common troubleshooting point. Potential issues include:
- Improper Localization: The prey protein may not be localized to the same cellular compartment (e.g., the plasma membrane) as your bait protein.
- Tag Interference: The protein tags (e.g., split-ubiquitin components) might be sterically hindering the interaction.
- Protein Misfolding: The heterologous expression in yeast might be causing misfolding of either the bait or prey protein.
- Technical Validation: Always ensure you have performed the necessary bait validation steps, as outlined in the MYTH protocol, to confirm your bait is correctly expressed and localized [99].
Comparative Analysis of Key Detection Techniques
The table below summarizes the core characteristics of several prominent methods for detecting membrane protein interactions.
Table 1: Comparison of Membrane Protein Interaction Detection Techniques
| Technique | Primary Mechanism | Throughput | Precision / Resolution | In Vivo Compatibility | Key Advantages | Key Limitations |
|---|---|---|---|---|---|---|
| Micropatterning on Surfaces [100] | Redistribution of fluorescent prey onto antibody-patterned surfaces. | High | Single-molecule sensitivity; Quantitative. | Live cells (compatible). | High sensitivity; detects weak interactions; quantitative data. | Requires specific antibody against bait; may not be suitable for all membrane protein types. |
| Membrane-SPINE [97] | In vivo crosslinking, affinity purification, and mass spectrometry. | Low to Medium | Identifies direct and indirect partners. | Yes (performed in living cells). | Captures transient interactions; works in native cellular context. | Lower throughput; requires optimization of crosslinking. |
| Membrane Yeast Two-Hybrid (MYTH) [99] | Split-ubiquitin system reconstitution in yeast. | High | Identifies binary interactions. | Yes (in yeast host). | Well-established for large-scale screening; uses full-length membrane proteins. | Interactions occur in heterologous system (yeast); potential for false positives. |
| Computational Prediction (PLM-interact) [101] [102] | Protein language models trained on sequence and evolutionary data. | Very High | Molecular detail (emerging); high AUPR on benchmarks. | N/A ( in silico ) | Extremely fast and scalable; no experimental reagents needed. | Performance drops for evolutionarily distant proteins; requires experimental validation. |
| Affinity Selection Mass Spectrometry (AS-MS) [103] | Incubation of target with compound library, isolation of binders, and MS analysis. | High | Identifies direct small-molecule binders. | No ( in vitro with purified protein). | Excellent for drug screening; directly identifies bound ligands. | Requires purified, functional membrane protein. |
Detailed Experimental Protocols
Protocol 1: Membrane-SPINE forIn VivoCross-Linking and Identification
Membrane-SPINE (Strep-protein interaction experiment) is a biochemical tool to identify in vivo protein-protein interactions of membrane proteins, even for transient ones [97].
Detailed Methodology:
- Cell Culture and Cross-linking:
- Grow bacterial cells (or other cell types) expressing the membrane bait protein with a C-terminal Strep-tag.
- Under a safety fume hood, split the culture. Add formaldehyde to a final concentration of 0.6% to the test sample for 20 minutes to cross-link interacting proteins. Keep a control sample without cross-linking.
- Centrifuge to collect cells and properly dispose of the formaldehyde-containing supernatant.
Membrane Protein Preparation:
- Resuspend the cell pellet in Tris-sucrose buffer with protease inhibitors.
- Add lysozyme in EDTA buffer to generate spheroplasts. Incubate on ice for 30 minutes.
- Collect spheroplasts by centrifugation and freeze the pellet at -20°C overnight.
- The next day, resuspend the pellet in a suitable buffer and sonicate on ice to disrupt the spheroplasts.
- Pellet the membrane fraction via ultracentrifugation at 100,000 x g for 30 minutes.
Solubilization and Affinity Purification:
- Resuspend the membrane pellet and solubilize the proteins using 2% Triton X-100 for one hour on ice.
- Ultracentrifuge again to remove insoluble material.
- Apply the supernatant to a Strep-Tactin column. Wash the column extensively with buffer.
- Elute the bait-prey complexes using a buffer containing desthiobiotin.
Reversal of Cross-links and Analysis:
- Split the eluate and boil one aliquot at 95°C for 20 minutes to reverse the formaldehyde cross-links.
- Analyze the samples by SDS-PAGE.
- Use immuno-blotting with prey-specific antibodies or mass spectrometry to identify co-purified interaction partners. A specific band appearing only in the cross-linked and boiled sample indicates a direct interaction.
Protocol 2: Micropatterning for Quantitative Analysis in Living Cells
This method allows for analyzing interactions between a fluorophore-labeled "prey" protein and a membrane-anchored "bait" protein in living cells by visualizing prey redistribution onto micropatterned surfaces [100].
Detailed Methodology:
- Surface Patterning:
- Create a micropatterned surface functionalized with antibodies specific to the exoplasmic domain of the bait membrane protein.
Cell Preparation and Plating:
- Transfer cells expressing both the bait membrane protein and a fluorescently-labeled prey protein onto the micropatterned surface.
Interaction Assay and Imaging:
- If the bait and prey interact, the fluorescent prey will co-redistribute and accumulate on the antibody-based micropatterns.
- This redistribution is quantified using fluorescence microscopy. The technique is sensitive enough to detect interactions at the single-molecule level and can be adapted for high-throughput screening.
Protocol 3: Computational Prediction with PLM-interact
For large-scale screening, computational methods like PLM-interact offer a powerful, sequence-based approach [101].
Detailed Methodology:
- Input Preparation:
- Provide the amino acid sequences of the two query proteins in FASTA format.
Model Inference:
- The PLM-interact model, which is based on a fine-tuned ESM-2 protein language model, jointly encodes the protein pair.
- It uses a transformer architecture with a "next sentence prediction" task to learn the relationship between the two sequences, determining if they are likely to interact.
Output Analysis:
- The model outputs a probability score indicating the likelihood of a physical interaction.
- A threshold (e.g., 0.5) can be applied to classify the pair as interacting or non-interacting. The model can also be fine-tuned to predict the effect of mutations on interactions.
Workflow Visualization
The following diagram illustrates the fundamental difference between the workflows of a key experimental method (Membrane-SPINE) and a computational method (PLM-interact).
Diagram 1: Comparison of experimental and computational workflows for detecting membrane protein interactions.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for Membrane Protein Interaction Studies
| Reagent / Material | Function in Experiment | Key Considerations |
|---|---|---|
| Formaldehyde [97] | Reversible cross-linker to trap transient protein-protein interactions in vivo. | Concentration and incubation time must be optimized to minimize non-specific cross-linking. Highly toxic; use in a fume hood. |
| Strep-Tactin Column [97] | Affinity purification matrix for isolating the Strep-tagged bait protein and its cross-linked partners. | Provides high purity and specificity under mild, non-denaturing conditions. |
| Triton X-100 [97] | Non-ionic detergent used to solubilize membrane proteins from the lipid bilayer. | Can disrupt some protein-protein interactions; milder alternatives (e.g., DDM) may be tested. |
| pMCSG53 Vector [104] | Bacterial expression vector with an N-terminal, cleavable hexa-histidine tag for recombinant protein expression. | Common starting point for high-throughput pipelines to produce soluble proteins for in vitro studies. |
| Styrene-Maleic Acid (SMA) Copolymer [98] | A detergent-free alternative to solubilize membrane proteins directly into native nanodiscs (SMALPs). | Preserves the native lipid environment and protein complex integrity, which is crucial for functional studies. |
| ESM-2 (650M parameter model) [101] | A large protein language model that serves as the foundation for the PLM-interact prediction tool. | Requires significant computational resources for training and fine-tuning, but not for basic inference. |
Membrane proteins are critical drug targets, but their inherent hydrophobicity, low natural abundance, and complex lipid bilayer environment make the detection and characterization of their interactions notoriously challenging [103] [105]. A persistent theme in the literature is that the limitations inherent to any single biochemical technique mean that relying on one method often yields an incomplete, and sometimes misleading, picture. This technical support center is built upon the core thesis that an orthogonal approach—the use of multiple, complementary methodologies—is not merely beneficial but essential for generating robust, reproducible, and physiologically relevant data on membrane protein interactions. The following guides and FAQs are designed to help you troubleshoot common experimental pitfalls by strategically integrating different techniques to validate your findings.
Frequently Asked Questions (FAQs) & Troubleshooting Guides
FAQ 1: My immunoprecipitation experiments for a membrane protein complex yield inconsistent results. How can I confirm if the interaction is real or an artifact?
Challenge: Traditional co-immunoprecipitation (co-IP) can be plagued by false positives from non-specific binding or weak, transient interactions that are lost during stringent washing steps. For membrane proteins, the use of detergents for solubilization can disrupt native complexes or introduce artifactual interactions [106].
Orthogonal Solution Strategy:
- Validate with Proximity Labeling (PL): Employ techniques like TurboID or APEX2 fused to your protein of interest. These enzymes covalently tag proximal proteins with biotin in living cells, capturing transient interactions in a near-physiological environment before cell lysis [106]. The biotinylated partners are then purified under denaturing conditions, drastically reducing false positives from post-lysis association.
- Correlate with Structural Data: If feasible, use cryo-Electron Tomography (cryo-ET) on isolated organelles or focused ion beam (FIB)-milled cells. This can visually confirm the spatial association of protein complexes within the native membrane, as demonstrated for lysosomal membrane proteins like V-ATPase and Clathrin [107].
Troubleshooting Guide:
- Problem: High background in PL experiments.
- Solution: Optimize biotin incubation time and concentration. For TurboID, shorter times (e.g., 10-30 minutes) can minimize background. Use peptide-level enrichment for mass spectrometry to directly identify biotinylation sites and increase specificity [106].
- Problem: Interaction is not detected by PL.
- Solution: The interaction might be very transient or require specific activation. Consider using a stimulation step (e.g., ligand addition) during the biotinylation window. Alternatively, the epitope tag or the enzyme fusion might be interfering with the interaction; test different fusion orientations (N- or C-terminal).
FAQ 2: How can I map the topology of a novel multi-pass transmembrane protein without high-resolution structural data?
Challenge: Computational predictors are not always accurate, and high-resolution techniques like crystallography may not be feasible. Engineered reporter tags (e.g., glycosylation sites) can sometimes disrupt folding or membrane integration [105].
Orthogonal Solution Strategy:
- Native Epitope Accessibility Mapping: Use a simple immunofluorescence protocol on live, non-permeabilized cells. The accessibility of native epitopes to antibodies is determined by confocal microscopy or flow cytometry. Antibody binding in intact cells indicates an extracellular domain, while binding only in permeabilized cells indicates an intracellular domain. Plasma membrane integrity must be monitored with a cell-impermeable probe like propidium iodide [105].
- Corroborate with AlphaFold2 and Peptide-Level PL: Utilize AlphaFold2 for a computational structural prediction to guide experimental design [105]. Furthermore, peptide-level analysis from PL data can help infer membrane protein topology by identifying which specific residues (e.g., on a cytoplasmic loop) are biotinylated, providing experimental evidence for the location of protein domains [106].
Troubleshooting Guide:
- Problem: No antibody binding to native epitopes on live cells.
- Solution: This does not definitively prove an intracellular location. The epitope may be masked by glycosylation or other steric hindrances. Always confirm with a positive control (e.g., a known extracellular domain of another protein) and use the permeabilized cell data as the definitive readout for intracellular localization.
- Problem: AlphaFold2 prediction conflicts with experimental data.
- Solution: Trust the experimental data. AlphaFold2 predictions are static and may not account for unique lipid-protein interactions or dynamic folding processes in the native membrane [105].
FAQ 3: My target is a G Protein-Coupled Receptor (GPCR). How can I perform high-throughput ligand screening and simultaneously identify allosteric binders?
Challenge: GPCRs are dynamic membrane proteins. Standard functional assays may miss allosteric modulators that do not directly block the orthosteric site but modulate receptor activity.
Orthogonal Solution Strategy:
- Affinity Selection-Mass Spectrometry (AS-MS) Screening: Incubate the purified GPCR with a large library of small molecules. Use size-exclusion chromatography or other methods to separate ligand-receptor complexes from unbound compounds, then use MS to identify the bound ligands. This is a powerful, label-free method for high-throughput screening [103] [108].
- Competition AS-MS for Mechanism of Action: To distinguish allosteric from orthosteric binders, perform competition AS-MS. Pre-incubate the receptor with a known orthosteric ligand, then add the library. Ligands that still bind in the presence of the orthosteric ligand are likely acting at an allosteric site [108].
- Functional Validation with Native MS: For promising hits, use native MS to directly analyze the intact GPCR-ligand complexes. This technique can preserve non-covalent interactions in a near-native lipid environment, allowing you to observe stoichiometry and proteoform-specific effects, providing direct mechanistic insights [103].
Troubleshooting Guide:
- Problem: Low hit rate in AS-MS screening.
- Solution: Ensure the receptor is properly folded and functional in the detergent or nanodisc system used. Optimize the buffer conditions to maintain protein stability and reduce non-specific binding [108].
- Problem: Difficulty detecting weak binders.
- Solution: AS-MS is highly sensitive, but very weak or fast-dissociating interactions can be missed. Complement it with surface plasmon resonance (SPR) or other biophysical techniques that can better characterize binding kinetics.
Comparative Methodologies at a Glance
The table below summarizes key techniques, enabling you to quickly compare their applications and limitations.
Table 1: Orthogonal Techniques for Membrane Protein Interaction Analysis
| Technique | Key Principle | Key Advantage | Primary Limitation | Ideal for: |
|---|---|---|---|---|
| Cryo-Electron Tomography (cryo-ET) [107] | Direct 3D visualization of frozen-hydrated cellular samples in situ. | Reveals native architecture and spatial organization of protein complexes in membranes. | Low throughput; technically demanding; requires specialized equipment. | Visualizing protein complexes in organelles, confirming spatial relationships. |
| Proximity Labeling (e.g., TurboID) [106] | Enzyme-catalyzed, covalent biotin-tagging of proximal proteins in live cells. | Captures weak/transient interactions in physiological context; no detergent lysis artifacts. | Potential for background labeling; enzyme size may sterically hinder. | Mapping entire interactomes of a target protein in its native cellular environment. |
| Affinity Selection-MS (AS-MS) [103] [108] | Physically separating (via size, affinity) ligand-protein complexes for MS identification. | Label-free, high-throughput screening of compound libraries against purified targets. | Requires purified, functional protein; may miss very weak binders. | High-throughput drug screening, fragment-based lead discovery. |
| Native Mass Spectrometry [103] | Analysis of intact protein complexes under non-denaturing conditions. | Directly observes ligand binding, stoichiometry, and lipid interactions. | Requires careful sample preparation and specialized instrumentation. | Mechanistic studies of drug binding and off-target effects. |
| Bacterial Two-Hybrid (B2H) [109] | Reconstitution of transcription factors via bait-prey interaction in E. coli. | Cost-effective, scalable; suitable for membrane protein interactions. | Limited to binary interactions; may lack eukaryotic PTMs. | Initial screening and validation of binary PPIs, particularly for bacterial proteins. |
| Advanced Light Microscopy (SMLM) [110] | Localization of single molecules with super-resolution via photochemical switching. | Provides single-molecule resolution of protein organization in the live cell membrane. | Limited throughput; requires specialized fluorophores and analysis. | Studying nanoscale clustering and lateral organization of membrane proteins. |
Essential Experimental Workflows
Workflow 1: Integrated In-Situ Interaction Mapping
This workflow combines cellular biochemistry with structural visualization for high-confidence interaction mapping.
Workflow 2: Ligand Screening and Validation Pipeline
This workflow is designed for drug discovery, from initial screening to mechanistic validation.
Research Reagent Solutions
The following table lists key reagents essential for implementing the orthogonal approaches discussed.
Table 2: Essential Research Reagents and Their Applications
| Reagent / Tool | Function | Key Application |
|---|---|---|
| Rho1D4 Antibody & 1D4 Peptide [107] | Immunopurification of epitope-tagged membrane proteins. | Gentle elution with 1D4 peptide for isolating intact organelles for cryo-ET. |
| TurboID / APEX2 Enzymes [106] | Genetically encodable proximity labeling catalysts. | Mapping protein interactomes in live cells and animals with high temporal resolution. |
| Styrene-Maleic Acid (SMA) Copolymer [8] | Directly solubilizes membranes to form native nanodiscs (SMALPs). | Extracting membrane proteins with their native lipid annulus for functional and structural studies. |
| Biotin-Phenol [106] | Substrate for APEX/APEX2 peroxidase enzymes. | Converted to a radical that labels nearby proteins upon addition of H₂O₂. |
| Nanodiscs (MSP / Saposin) [8] | Forms a controllable lipid bilayer disc to stabilize membrane proteins. | Creating a near-native lipid environment for purifying proteins for AS-MS, Native MS, or biochemistry. |
| Propidium Iodide [105] | Cell-impermeable fluorescent DNA dye. | A critical control to monitor plasma membrane integrity in live-cell topology mapping assays. |
Membrane proteins constitute 20–30% of all proteins in sequenced genomes and represent nearly 50% of modern drug targets [111]. However, their inherent hydrophobicity, complex topology within lipid bilayers, and frequent involvement in transient interactions make them notoriously difficult to study. Transient protein-protein interactions are often weak, with dissociation constants in the micromolar range, and short-lived, lasting seconds or less [112]. No single experimental method can fully capture the complexity of their dynamics, stoichiometry, and structural basis. This technical support center outlines an integrated framework, combining Co-IP, FRET, and computational modeling, to overcome these limitations and provide a holistic view of membrane protein interactions for drug discovery research.
Troubleshooting Guides
Co-Immunoprecipitation (Co-IP) Troubleshooting
Co-IP is a cornerstone technique for detecting direct protein-protein interactions under near-physiological conditions [113]. However, when applied to membrane proteins, several specific challenges arise.
FAQ: My Co-IP shows a strong bait protein band but weak or no prey protein bands. What could be wrong?
- Cause: The lysis conditions may be too stringent, disrupting native protein-protein interactions. RIPA buffer, which contains ionic detergents like sodium deoxycholate, is known to denature proteins and prevent interactions, making it unsuitable for Co-IP of membrane complexes [114].
- Solution: Use a milder, non-denaturing lysis buffer (e.g., Cell Lysis Buffer #9803) [114]. Ensure the buffer contains mild non-ionic detergents (e.g., Triton X-100) to solubilize membrane proteins while preserving interactions [115] [113]. Include protease inhibitors and perform all steps on ice to prevent protein degradation [116].
FAQ: I get a high background and non-specific bands in my Co-IP results. How can I reduce this?
- Cause: Non-specific binding of proteins to the beads or the antibody itself [114] [116].
- Solution:
- Pre-clearing: Incubate your lysate with beads alone for 30-60 minutes at 4°C before adding the antibody [114].
- Optimize washes: Increase the concentration of detergent (e.g., Tween-20) and salt (NaCl) in your wash buffer to dissociate weak, non-specific interactions [116] [113].
- Include critical controls: Always run a bead-only control and an isotype control (an antibody of the same species but irrelevant specificity) to identify the source of background [114] [57].
FAQ: My input control shows the protein is present, but I cannot pull it down. What should I check?
- Cause: The antibody's epitope on the target (bait) protein might be masked by the protein's conformation or an interacting partner [114].
- Solution: Try an antibody that recognizes a different epitope on the target protein. Alternatively, use a tagged version of the bait protein and an antibody against the tag [114] [115].
FRET Assay Troubleshooting
FRET (Förster Resonance Energy Transfer) acts as a "molecular ruler," providing nanometer-scale distance information ideal for studying membrane protein associations in live cells [37].
FAQ: How can I study membrane receptor oligomers with FRET when the exact oligomer size is unknown?
- Challenge: Traditional FRET analysis struggles to define oligomer size beyond dimers [117].
- Solution: Implement an effective equilibrium dissociation constant. This constant denotes the receptor concentration at which half are monomeric and inactive, and half are associated into active oligomers, irrespective of the exact size. This provides a well-defined measure of oligomer stability and activity from FRET data, even for heterogeneous distributions [117].
FAQ: What are the key advantages of advanced FRET variants for studying PPIs?
- Time-Resolved FRET (TR-FRET): Uses long-lifetime probes (e.g., lanthanide chelates) and time-gated detection to eliminate short-lived background fluorescence, significantly enhancing sensitivity for low-abundance targets [37].
- FLIM-FRET (Fluorescence Lifetime Imaging Microscopy-FRET): Measures changes in the donor's fluorescence lifetime, which is independent of fluorophore concentration, enabling direct visualization of PPIs with high temporal and spatial resolution [37].
- Single-Molecule FRET (smFRET): Observes individual molecules, revealing heterogeneous behavior, transient states, and dynamics that are averaged out in ensemble measurements [37].
FAQ: Why is my FRET signal low or inconsistent?
- Cause: Low expression of the target proteins or improper spectral unmixing.
- Solution: Ensure adequate expression levels of donor- and acceptor-tagged proteins. Use single transfections of donor-only and acceptor-only constructs to acquire proper basis spectra for unmixing the FRET emission spectra [117].
Computational Data Integration
Computational methods are indispensable for complementing experimental data, predicting structures, and modeling dynamics, especially where experimental characterization is difficult [111] [118].
FAQ: How can I obtain structural insights for my membrane protein of interest if there is no experimental structure?
- Solution: Utilize computational modeling tools. Machine-learning (ML) based structure prediction algorithms (like AlphaFold2) can generate highly accurate 3D models. Subsequently, Molecular Dynamics (MD) simulations can be used to study the dynamics of the predicted model within a lipid bilayer environment, providing insights into conformational changes and interaction interfaces [111].
FAQ: My Co-IP suggests an interaction, but I am unsure if it is direct or mediated by a third party. How can I investigate this?
- Solution: Structure-based in silico approaches can predict whether two proteins are likely to interact directly based on the complementarity of their surfaces. Docking simulations can model the atomic-level details of the proposed complex, providing a testable hypothesis for direct binding [118].
Integrated Workflow: A Practical Protocol
To effectively study a membrane protein interaction, follow this sequential, integrated protocol.
Stage 1: Initial Validation and Complex Pull-Down with Co-IP
- Cell Lysis: Harvest and lyse cells expressing your membrane protein of interest using a mild, non-ionic lysis buffer (e.g., containing Triton X-100) supplemented with protease and phosphatase inhibitors [114] [113].
- Pre-clearing: Incubate the lysate with Protein A/G beads for 1 hour at 4°C to reduce non-specific binding. Centrifuge and collect the supernatant [115].
- Immunoprecipitation: Incubate the pre-cleared lysate with a specific antibody against your target bait protein for several hours or overnight at 4°C. Then add Protein A/G beads and incubate further to capture the antibody-protein complex [115] [113].
- Washing and Elution: Wash the beads thoroughly with lysis buffer containing increased salt and detergent to remove non-specifically bound proteins. Elute the bound protein complex using a gentle, low-pH glycine buffer or by boiling in SDS-PAGE loading buffer [113].
- Analysis: Analyze the eluate by Western blotting to confirm the presence of suspected interacting partners.
Stage 2: Confirming Direct Interaction and Stoichiometry with FRET
- Construct Design: Create plasmids encoding your membrane proteins of interest tagged with a FRET donor (e.g., mTurquoise) and acceptor (e.g., eYFP) [117].
- Cell Transfection: Co-transfect the donor- and acceptor-tagged constructs into your target cells (e.g., HEK293T). Include controls transfected with donor-only and acceptor-only constructs [117].
- FRET Imaging: Acquire images of live cells using a sensitive microscope (e.g., a two-photon or confocal microscope with spectral imaging capabilities). Perform scans at both donor and acceptor excitation wavelengths [117].
- Data Analysis: Unmix the emission spectra into donor and acceptor components using the control spectra. Calculate the apparent FRET efficiency (
Eapp) and the concentrations of donor ([D]) and acceptor ([A]) in the plasma membrane. For oligomeric receptors, plotEappagainst receptor concentration to derive the effective dissociation constant [117].
Stage 3: Structural and Dynamic Modeling with Computational Tools
- Structure Prediction: Input the amino acid sequence of your membrane protein into a structure prediction server (e.g., AlphaFold Protein Structure Database) to obtain a 3D model [111].
- System Setup: Embed the predicted model into a simulated lipid bilayer that mimics the native membrane environment. Solvate the system in an appropriate water model and add ions to physiological concentration.
- Molecular Dynamics Simulation: Run an all-atom MD simulation to observe the dynamics and stability of the protein or protein complex over time (nanoseconds to microseconds). Analyze the trajectory to identify key residues at the interaction interface and characterize the conformational flexibility [111].
Data Presentation and Analysis
Table 1: Comparison of PPI Technique Capabilities for Membrane Proteins
Table summarizing the strengths and limitations of different methods.
| Technique | Under Physiological Conditions? | Can Detect Transient PPIs? | Provides Dynamic/Kinetic Info? | Spatial Resolution | Key Limitations for Membrane Proteins |
|---|---|---|---|---|---|
| Co-IP [57] [37] [118] | Yes (in lysates) | Partially (can be enhanced with crosslinkers) | No | Low (ensemble) | - Lysis conditions can disrupt complexes.- Captures indirect interactions.- Provides a snapshot, no dynamics. |
| FRET [117] [37] | Yes (in live cells) | Yes | Yes | Excellent (nanometer) | - Can be challenging to quantify for >dimers.- Requires labeling, which may perturb function. |
| Yeast Two-Hybrid (Y2H) [37] [118] | No (nuclear) | Limited | No | Low | - Unsuitable for many membrane proteins.- High false positive rate. |
| Computational Modeling [111] | In silico | Yes (via MD) | Yes (via MD) | Excellent (atomic) | - Predictions require experimental validation.- Computationally expensive. |
Table 2: Research Reagent Solutions for Integrated Membrane Protein Studies
Essential materials and their functions for the featured experiments.
| Item | Function | Application Notes |
|---|---|---|
| Mild Lysis Buffer (e.g., with Triton X-100) | Solubilizes membrane proteins while preserving non-covalent protein-protein interactions [114] [113]. | Critical for Co-IP; avoid strong denaturing buffers like RIPA for interaction studies. |
| Protein A/G Beads | Binds the Fc region of antibodies to capture and pull down the antigen-antibody complex [114] [113]. | Choose Protein A for rabbit IgG, Protein G for mouse IgG for higher affinity [114]. |
| Crosslinkers (e.g., DSS, BS3) | Covalently stabilizes weak or transient protein interactions immediately in cells or lysates, "freezing" them for analysis [57] [113]. | Membrane-permeable (DSS) for intracellular targets; membrane-impermeable (BS3) for cell surface proteins. |
| FRET Donor/Acceptor Pair (e.g., mTurquoise/eYFP) | Genetically encoded tags that enable FRET-based proximity measurement (1-10 nm) in live cells [117] [37]. | mTurquoise is a bright, photostable donor; eYFP is a common acceptor. Linker length should be optimized. |
| Protease/Phosphatase Inhibitor Cocktail | Prevents degradation and dephosphorylation of proteins and their modifications during sample preparation [114]. | Essential for maintaining post-translational modifications which often regulate membrane protein function. |
Visualized Workflows and Pathways
Diagram 1: Integrated Workflow for Membrane Protein Interaction Analysis
This flowchart outlines the sequential and iterative process of integrating Co-IP, FRET, and computational modeling to build a comprehensive model of membrane protein interaction.
Diagram 2: Co-IP and FRET Experimental Relationships
This diagram illustrates the complementary questions answered by Co-IP, FRET, and computational modeling, highlighting their respective strengths and limitations in a unified framework.
Identifying True vs. Indirect Interactions in Large Complexes
### FAQs on Differentiating Protein Interactions
Q1: What are the primary experimental challenges in distinguishing direct from indirect membrane protein interactions? Membrane proteins present unique challenges, including their structural complexity, low native expression levels, and the disruption of their native lipid environment during purification. Traditional detergent-based extraction can strip away essential co-factors and lipids, destabilizing the protein and obscuring true interactions. Furthermore, incomplete proteolytic digestion and poor compatibility with standard mass spectrometry (MS) protocols make it difficult to capture intact, functional complexes for analysis [79].
Q2: How can I confirm that a detected interaction is direct and not part of a larger, indirect complex? A multi-method approach is crucial for verification:
- FRET (Förster Resonance Energy Transfer): Use FRET to detect nanometer-scale proximity between labeled proteins. A high FRET efficiency indicates direct interaction or very close association [119].
- Fluorescence Cross-Correlation Microscopy (FCCM): This technique directly measures the co-diffusion of two differently labeled proteins through a small observation volume. Coordinate bursts of fluorescence confirm they are part of the same complex in living cells [119].
- Stability-based MS assays: Techniques like native MS or solution-phase thermochemistry, when combined with membrane mimetics, can help preserve and identify the specific subunits within a stable, intact complex, helping to differentiate core interactions from peripheral ones [79].
Q3: My data from FRET and BiFC experiments is conflicting. How should I interpret this? FRET and Bimolecular Fluorescence Complementation (BiFC) report on different aspects of an interaction. FRET is sensitive to close proximity and is reversible, making it suitable for detecting both stable complexes and transient collisions. In contrast, BiFC involves the irreversible re-folding of two fluorescent protein fragments; it is excellent for confirming that an interaction can occur but is less suited for studying its dynamics or reversibility. Conflicting data may arise if an interaction is transient (detected by FRET) but not stable enough to form a fluorescent BiFC complex, or if the BiFC fragments reassemble slowly, missing rapid interaction cycles [119].
Q4: Which computational tools can help predict direct protein-protein interactions, especially when experimental structures are unavailable? SpatialPPIv2 is a graph-neural-network-based model designed for this purpose. It uses protein language models to embed sequence features and graph attention networks to capture structural information. A key advantage is that it does not depend on experimentally determined structures and can use predicted structures from AlphaFold2, AlphaFold3, or ESMFold, maintaining robustness even in the absence of high-resolution structural data [120].
### Troubleshooting Guides
Issue: High Background Noise in FRET Measurements Low FRET efficiency or high background can stem from multiple factors. Follow this diagnostic pathway to identify and correct the problem:
Table: Troubleshooting Steps for FRET Background Noise
| Step | Action | Expected Outcome |
|---|---|---|
| 1. Check Fluorophore Pair | Verify use of optimal FRET pairs (e.g., mCerulean/mVenus or CyPet/SYFP2). Ensure spectral overlap is high [119]. | Increased FRET efficiency signal. |
| 2. Assess Labeling | Confirm high labeling efficiency. If using fluorescent proteins (FPs), check for proper fusion and folding. Consider small biarsenical tags (FlAsH/ReAsH) to reduce steric hindrance [119]. | Reduced population of unpaired donors/acceptors. |
| 3. Control for Expression | Verify correct subcellular localization and absence of protein aggregation. Compare to negative control cells expressing only the donor or acceptor [119]. | Elimination of signal from mislocalized or overexpressed proteins. |
| 4. Quantify Cross-talk | Perform control measurements to calculate and subtract spectral bleed-through from the FRET signal. | A cleaner, more accurate FRET efficiency measurement. |
Issue: Low Recovery of Intact Membrane Protein Complexes for MS Analysis The failure to recover intact complexes often relates to the method of solubilization and the maintenance of a native-like environment.
Table: Strategies for Stabilizing Membrane Protein Complexes
| Challenge | Solution | Mechanism |
|---|---|---|
| Detergent Destabilization | Use tailored detergent architectures or MS-compatible detergents [79]. | Minimizes denaturation and preserves functional conformations. |
| Loss of Native Lipid Environment | Incorporate complexes into Membrane Mimetics (MMs) like nanodiscs, peptidiscs, or styrene-maleic acid (SMA) copolymers [79]. | Provides a native-like lipid bilayer that maintains ligand-binding sites and protein-lipid interactions. |
| Complex Disassembly in Solution | Apply stability-based MS assays (e.g., native MS, ion mobility-MS) [79]. | Allows analysis of intact assemblies under non-denaturing conditions, capturing co-bound lipids and ligands. |
### Research Reagent Solutions
Table: Essential Reagents for Differentiating Protein Interactions
| Reagent / Tool | Function / Application |
|---|---|
| Monomeric FPs (mCerulean, mVenus) | Genetically encoded tags for FRET and BiFC experiments with minimal oligomerization [119]. |
| Tetracysteine Motif Tags (FlAsH, ReAsH) | Small biarsenical fluorophores for labeling with reduced steric interference compared to FPs [119]. |
| Membrane Mimetics (Nanodiscs, SMA Polymers) | Provide a native-like lipid environment to stabilize membrane proteins for MS and functional studies [79]. |
| SpatialPPIv2 Software | A computational tool for predicting direct PPIs using sequence and structural features, even with predicted models [120]. |
| Fluorescence Correlation Spectroscopy (FCM) Setup | Instrumentation for measuring diffusion coefficients and stoichiometry of complexes in living cells [119]. |
Benchmarking Computational Predictions Against Experimental Gold Standards
Troubleshooting Common Experimental-Computational Discrepancies
My computational model predicts strong protein-protein interactions, but my FRET experiments show no signal. What could be wrong?
Several factors could cause this discrepancy. Your fluorescent protein tags (e.g., GFP, YFP) may be improperly folded or sterically hindered from interacting, especially with large tags like GFP. The linkers between your membrane protein and the fluorescent tag might be too rigid or too short, preventing the fluorophores from coming close enough for energy transfer. Additionally, the local cellular environment, such as membrane curvature or lipid composition, might differ between your computational model's assumptions and the experimental conditions. We recommend verifying fluorophore functionality with positive controls, testing different linker lengths, and ensuring your computational model accounts for the full fusion protein structure, not just the native protein [12].
My super-resolution microscopy reveals protein clustering, but my molecular dynamics simulations show uniform distribution. Why the mismatch?
This typically stems from limitations in either the simulation timescales or the model's completeness. Standard all-atom molecular dynamics simulations rarely exceed microsecond timescales, while biological clustering may occur over seconds or minutes. Your simulation might also lack critical cellular components known to drive clustering, such as specific lipids, cytoskeletal elements, or crowding agents. We suggest implementing enhanced sampling techniques in your simulations to accelerate rare events, incorporating realistic membrane compositions based on cryo-EM data, and applying spatial analysis techniques (like cluster analysis) to both your experimental and simulation data for direct comparison [12] [8].
How can I improve my membrane protein structural model for better computational predictions?
With recent advances in deep learning-based structure prediction, you now have powerful new options. Tools like AlphaFold2, AlphaFold3, and ESMFold can generate high-quality predicted structures, especially valuable for membrane proteins where experimental structures are scarce. Use these predicted structures as starting points for molecular dynamics simulations. Incorporate structural constraints from experimental techniques like cryo-EM, which can resolve lipid interactions, to guide and validate your models. Focus particularly on domains that these tools highlight as functionally important [121] [122].
The binding kinetics from my simulations don't match my experimental measurements. How can I resolve this?
Drug-target binding kinetics are notoriously challenging to simulate accurately due to the large timescale gap between simulation capabilities (microseconds) and biological reality (hours). The problem may lie in insufficient sampling of binding/unbinding pathways, force field inaccuracies for intermediate states, or overlooking allosteric regulation sites. We recommend using specialized enhanced sampling MD methods like metadynamics or Markov state models that are specifically designed for kinetics. Always benchmark your computational methods on systems with known experimental kinetics first, and collaborate closely with experimentalists to generate reliable benchmark data for your specific protein-ligand systems [123].
Performance Benchmarks: Computational Methods vs. Experimental Standards
Table 1: Benchmarking Protein Function Prediction Methods Using Experimental PDB Structures
| Method | Type | Molecular Function (Fmax) | Cellular Component (Fmax) | Biological Process (Fmax) | Key Features |
|---|---|---|---|---|---|
| DPFunc | Structure-based (Deep Learning) | 0.759 | 0.783 | 0.731 | Domain-guided attention, ESM-1b features, GCN on structures |
| GAT-GO | Structure-based (GNN) | 0.653 | 0.614 | 0.558 | Graph attention networks, ESM-1b features |
| DeepFRI | Structure-based (GNN) | 0.627 | 0.569 | 0.527 | Graph convolutional networks, protein contact maps |
| DeepGO | Sequence-based (Deep Learning) | 0.547 | 0.576 | 0.482 | Protein sequences and protein-protein interactions |
| Blast | Homology-based | 0.357 | 0.401 | 0.324 | Sequence similarity search |
Performance metrics (Fmax) are shown without post-processing. DPFunc demonstrates significant improvement across all Gene Ontology categories by incorporating domain-guided structure information [121].
Table 2: Experimental Techniques for Validating Computational Predictions
| Experimental Method | Applications in Validation | Key Measurable Parameters | Technical Considerations |
|---|---|---|---|
| FRET / FLIM | Protein-protein interactions, oligomerization | Distances (1-10 nm), interaction states, cluster size | Requires fluorophore labeling; can be affected by photophysics |
| Super-resolution microscopy (STED, PALM/dSTORM) | Spatial distribution, nanoscale organization | Cluster size, density, colocalization | Resolution beyond diffraction limit; complex sample prep |
| Fluctuation spectroscopy (FCS, RICS, SpIDA) | Oligomeric state, diffusion coefficients | Brightness, particle number, oligomer size | Requires high sensitivity detection; analysis complexity |
| Cryo-electron microscopy | High-resolution structure, lipid interactions | Atomic coordinates, lipid binding sites | Sample vitrification; membrane mimetics required |
| Ligand binding studies | Binding kinetics, affinity | Kon, Koff, KD, residence time | Requires functional reconstitution; label-free options available |
Experimental Protocols for Method Validation
Quantitative FRET Efficiency Measurements
This protocol validates computational predictions of membrane protein interactions by measuring energy transfer between fluorophores.
- Sample Preparation: Express your membrane protein fused to donor (CFP) and acceptor (YFP) fluorophores in Expi293F cells using the pEF6 V5-His TOPO TA expression vector for optimal yields [124].
- Cell Membrane Preparation: Harvest cells and isolate membrane fractions using differential centrifugation. Solubilize membranes with detergent (e.g., DDM at 100× CMC) or incorporate into nanodiscs to preserve native interactions [6].
- Data Acquisition: Acquire fluorescence emission spectra (excitation at 433 nm) from 450-600 nm. Collect time-resolved fluorescence decays for FLIM-FRET measurements.
- Control Measurements: Include donor-only and acceptor-only samples for background subtraction and correction factors.
- Data Analysis: Calculate FRET efficiency using either sensitized acceptor emission or donor fluorescence lifetime: E = 1 - (τDA/τD), where τDA and τD are donor lifetimes in presence and absence of acceptor, respectively.
- Interpretation: Compare measured distances with computational predictions. FRET efficiencies of 5-10% correspond to distances of ~7-8 nm, while 30-40% efficiencies indicate closer proximity (~4-5 nm) [12].
Spatial Distribution Analysis via dSTORM
This protocol validates computational predictions of membrane protein clustering at nanoscale resolution.
- Sample Labeling: Express your membrane protein with an extracellular tag. Label live cells with primary antibodies and photoswitchable dye-conjugated secondary antibodies (e.g., Alexa Fluor 647).
- Imaging Buffer Preparation: Prepare oxygen-scavenging imaging buffer containing 100 mM mercaptoethylamine, 5% glucose, 0.8 mg/mL glucose oxidase, and 40 μg/mL catalase in PBS.
- Microscopy Acquisition: Use TIRF illumination with 640 nm laser for activation and 642 nm laser for excitation. Collect 10,000-20,000 frames at 50-100 ms exposure time.
- Localization Reconstruction: Detect single-molecule events and determine their precise coordinates (x,y) with ~20 nm precision using Gaussian fitting.
- Cluster Analysis: Apply density-based clustering algorithms (DBSCAN) to identify protein clusters. Calculate cluster size, density, and nearest-neighbor distances.
- Validation: Compare cluster parameters with computational predictions. Note that computational models may need to simulate minutes of membrane dynamics to observe spontaneous clustering observed experimentally [12].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for Membrane Protein Interaction Studies
| Reagent / Tool | Function | Application Notes |
|---|---|---|
| C41(DE3) E. coli cells | Protein expression with reduced toxicity | Point mutations in lacUV5 promoter reduce transcription rate; gentler on host cells [6] |
| pEF6 V5-His TOPO TA vector | Mammalian expression | Contains EF-1α promoter; optimized for membrane protein yields in 293FT cells [124] |
| Expi293F cells | Mammalian expression system | High transfection efficiency; optimized for use with ExpiFectamine transfection reagent [124] |
| Nanodiscs | Membrane protein solubilization | Preserves native lipid environment; superior for functional assays but larger size [6] [8] |
| Detergents (DDM, OG) | Membrane protein extraction | Forms micelles; use at 100× CMC; creates homogenous samples for crystallography [6] |
| Cobalt-charged resin | Affinity purification | Increases purity over nickel-based resins with some trade-off in recovery [6] |
| Styrene-maleic acid copolymers | Native membrane extraction | Forms "SMALPs" - preserves native lipid environment without detergent [8] |
Method Selection Workflow
Experimental-Computational Integration Framework
Frequently Asked Questions (FAQs)
What are the most common reasons for failure in membrane protein expression for structural studies?
The most frequent issues include protein toxicity to host cells, improper folding in non-native membranes, and insufficient stability once extracted from the membrane. To address toxicity, switch from BL21 to specialized strains like C41(DE3), C43(DE3), or Lemo21(DE3) E. coli cells, which have point mutations that reduce transcription rates. For stability problems, try using minimal growth media like M9 minimal medium, which slows growth and may improve folding. Additionally, consider expressing homologs from other species, as subtle sequence differences can dramatically improve stability, or add solubility tags like GFP or lysozyme units to improve expression yield and stability [6].
How long should I wait for membrane protein extraction to occur?
Allow at least three hours for extraction, but overnight incubation typically yields better results. Contrary to intuition, extraction is often more efficient at 20-30°C than at 4°C due to increased thermal motion that facilitates the solubilization process. Always verify that the elevated temperature doesn't harm your specific protein sample before proceeding [6].
My membrane protein isn't binding to the affinity column during purification. What should I try?
This common problem occurs because solubilizing agents (detergents, nanodiscs) can hide affinity tags. Use loose resin instead of a static column and physically mix it with your sample for several hours to encourage binding. Alternatively, dilute your sample at least 2-fold to reduce the concentration of the solubilizing agent. If these fail, move your affinity tag to the opposite terminus or extend it (e.g., from 6× His to 12× His) to make it more accessible [6].
What computational metrics best indicate a reliable membrane protein structural model?
For function prediction, examine the Fmax scores across Molecular Function, Cellular Component, and Biological Process ontologies. Methods like DPFunc achieving Fmax scores above 0.70 across categories demonstrate strong correlation with experimental annotations. For structural accuracy, check residue-wise attention scores that highlight functionally important domains, and verify that predicted lipid-facing residues align with cryo-EM density maps when available. High-performance models typically incorporate both sequence-based features (from tools like ESM-1b) and domain-guided structural information [121] [8].
Technical Support Center
Frequently Asked Questions (FAQs)
FAQ 1: What is the "research bias" against understudied and membrane proteins, and why is it a problem?
The "research bias" describes how scientific efforts remain concentrated on a small subset of well-known proteins, overlooking many biomedically important targets. This creates a significant gap in our knowledge and tools. [125]
- Quantitative Evidence: An analysis of the entire PubMed database found that 54.5% of human proteins are scarcely researched, mentioned in fewer than 50 publications. In contrast, a mere 5000 well-studied proteins account for 95% of all scientific publications. [125]
- The Impact on Models: This bias is perpetuated in model development. Popular gene annotation databases, like Gene Ontology, exhibit high levels of inequality, with a Gini coefficient of 0.63. Machine learning models trained on these biased datasets inevitably learn to perform poorly on the under-represented proteins, compromising their generalizability. [125]
- Specific Bias Against Membrane Proteins: Membrane proteins are critically under-represented in standard protein-protein interaction (PPI) datasets. While they constitute about 30% of the genome and 60% of all drug targets [126], they are often missing from datasets derived from common methods like yeast two-hybrid (Y2H) and affinity capture-mass spectrometry (affinity capture-MS). [126] For example, affinity capture-MS data, which contributes over 25% of interactions in the BIOGRID database, has less than 10% of its interactions involving a membrane protein. [126]
Table 1: Metrics of Research Bias in Protein Studies
| Metric | Finding | Implication for Model Generalizability |
|---|---|---|
| Publication Concentration [125] | 95% of publications focus on only 5,000 proteins. | Training data lacks diversity, leading to poor performance on understudied targets. |
| Annotation Inequality (Gini Coefficient) [125] | Combined Gini coefficient of 0.63 across gene annotation databases. | Models have limited functional information for most proteins, hindering accurate prediction. |
| Membrane Protein Under-representation [126] | Only ~10% of interactions in affinity capture-MS data involve a membrane protein. | Models are blind to a large class of therapeutically relevant protein interactions. |
FAQ 2: My model shows high overall accuracy (>90%), but fails on new understudied viral proteins. What is wrong?
This is a classic sign of data leakage and overfitting, where a model learns biases and patterns specific to the training data rather than generalizable rules. Standard evaluation metrics can be misleading for understudied protein classes. [127]
- The Problem: When a dataset is imbalanced, with many examples from well-studied proteins and few from understudied ones, a model can achieve high accuracy by simply correctly predicting the majority class. It may not have learned meaningful rules for the minority class (understudied proteins). [127]
- Troubleshooting Steps:
- Audit Your Training Data: Check the distribution of your data. How many positive and negative examples exist for the protein family you are trying to generalize to? For understudied viruses, the lack of both positive and negative data is a major constraint. [127]
- Implement Robust Evaluation: Move beyond simple random train-test splits.
- Use a viral protein-specific evaluation framework that categorizes proteins into "majority" and "minority" classes based on their representation in the dataset. [127]
- Perform independent blind testing with a balanced dataset, where the number of positive and negative examples for each protein is equal. This can reveal true performance, often causing a dramatic drop in accuracy (e.g., to below 50%). [127]
- Re-examine Negative Sampling: The method used to generate non-interacting protein pairs (negative data) can introduce hidden biases. Curate and assess multiple types of negative sampling strategies to ensure your model is not learning these artifacts. [127]
FAQ 3: What experimental methods are suitable for validating interactions involving understudied membrane proteins?
Traditional high-throughput methods like standard Yeast Two-Hybrid (Y2H) are often unsuitable because they require proteins to localize to the nucleus, which can cause membrane proteins to misfold. [126] You need methods designed for the unique cellular environment of membrane proteins.
- Recommended Experimental Protocols:
- Split-Ubiquitin Yeast Two-Hybrid (SU-2HY): This is a modified version of Y2H specifically designed for integral membrane proteins. It uses a split ubiquitin system to detect interactions in a membrane context. [128] [126] One screen using this technique identified 1,985 putative interactions involving 536 membrane-associated proteins that were missed by other methods. [126]
- Protein-fragment Complementation Assay (PCA): This method detects interactions by reconstituting a functional protein from two fragments when the bait and prey proteins interact. It is performed in the natural cellular context, which diminishes bias against membrane proteins. [126]
- Surface-Enhanced Raman Spectroscopy (SERS): This label-free and non-destructive technique can analyze membrane proteins without removing them from their native environment. It provides a "molecular fingerprint" and can be used for real-time, in-situ monitoring of dynamic processes like electron transfer in membrane-bound respiratory chain proteins. [129]
- Innovative Surface Plasmon Resonance (SPR) Fixation: New fixation technologies use directional immobilization and stabilization strategies to anchor membrane proteins to the SPR chip while preserving their structure and function. This can increase SPR signal strength by 2-5 times and reduce required sample volume by 50-80%, making it viable for rare membrane proteins. [130]
Table 2: Comparison of Methods for Membrane Protein Interaction Analysis
| Method | Key Principle | Advantages for Membrane Proteins | Key Limitations |
|---|---|---|---|
| Split-Ubiquitin Y2H [128] [126] | Reconstitution of a transcription factor via split ubiquitin at the membrane. | Specifically designed for membrane environments; amenable to screening. | Still an artificial system; potential for false positives/negatives. |
| Protein-fragment Complementation (PCA) [126] | Reconstitution of a functional enzyme upon protein interaction. | Occurs in the natural cellular context. | Can be irreversible; may not detect transient interactions. |
| Surface-Enhanced Raman Spectroscopy (SERS) [129] | Enhancement of Raman scattering signals on metal nanostructures. | Label-free, non-destructive, and capable of in-situ dynamic monitoring. | Can produce complex data that requires AI/ML for interpretation. [129] |
| Innovative SPR Fixation [130] | Directional immobilization of tagged proteins on a chip surface. | Preserves protein activity; highly sensitive and quantitative. | Can be technically complex and costly to set up. |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Resources for Studying Understudied Protein Interactions
| Reagent / Resource | Function & Application | Key Consideration |
|---|---|---|
| STRING Database [125] | A database of known and predicted protein-protein interactions, both physical and functional. | Useful for determining the "studiedness" of a protein (number of known interactors). Use the "physical" subnetwork for highest confidence. [125] |
| cBioPortal / TCGA Data [131] [125] | Open-source portals providing genomics data (mutations, CNA, expression) from thousands of tumor samples. | Critical for establishing the biomedical importance of an understudied protein via its link to disease. [125] |
| MalaCard [125] | An integrated database of human maladies compiled from multiple sources. | Used to find gene-disease links, another key metric for prioritizing understudied proteins. [125] |
| Directionally Tagged Membrane Proteins | For techniques like SPR, membrane proteins are engineered with tags (e.g., His-tag) for oriented immobilization. | Maximizes exposure of active sites and preserves functionality, leading to more accurate binding data. [130] |
| SERS-Active Nanostructures [129] | Metal nanoparticles or structured surfaces that enhance Raman signals for sensitive detection. | Enables the detection of low-concentration molecules and the study of membrane proteins in near-native conditions. [129] |
| AI/ML Cascade Algorithms [129] | Machine learning models designed to process and interpret complex spectral or sequence data. | For example, an MLC algorithm can automate the analysis of Raman spectra to assess PD-L1 expression levels, avoiding artifacts from traditional staining. [129] |
Experimental Workflow & Model Evaluation Diagrams
Workflow for Robust Model Development
Sources of Bias Against Membrane Proteins
Conclusion
The reliable detection of membrane protein interactions demands a multifaceted strategy that acknowledges the inherent limitations of any single method. Success hinges on selecting techniques appropriate for the interaction's dynamics—be it transient or stable—and rigorously validating findings through orthogonal approaches. The future of this field lies in the intelligent integration of advanced biophysical tools like cryo-EM and FLIM-FRET with powerful computational models, including deep learning. This synergistic approach, which combines high-resolution structural data with dynamic live-cell analysis and predictive power, is poised to break through current technological barriers. Such progress will not only deepen our fundamental understanding of cellular communication but also accelerate the discovery of novel therapeutic targets rooted in the membrane protein interactome, particularly for complex diseases like cancer and neurodegenerative disorders.
References
- 1. Immunoprecipitation Troubleshooting Guide [https://www.cellsignal.com/learn-and-support/troubleshooting/ip-troubleshooting-guide?srsltid=AfmBOooj65kiEB62WVEQDi_xeUClyTpnZDxR--0HlLtUQS0Uijr6XyMK]
- 2. Western blot troubleshooting guide! [https://www.jacksonimmuno.com/secondary-antibody-resource/technical-tips/western-blot-trouble-shooting/]
- 3. Editorial: Novel insights into the modulation of protein ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2025.1607512/full]
- 4. Regulating Membrane Proteins via Lipid Solvation [https://bioengineer.org/regulating-membrane-proteins-via-lipid-solvation/]
- 5. Molecular basis for the regulation of membrane proteins ... [https://pubmed.ncbi.nlm.nih.gov/41006774/]
- 6. Working with Membrane Proteins: 12 Simple Tips for Success [https://bitesizebio.com/52681/working-with-membrane-proteins/]
- 7. How to Make Membrane Protein Research Less Frustrating [https://www.fidabio.com/blogs/how-to-make-membrane-protein-research-less-frustrating]
- 8. Interactions between membrane proteins and lipid ... [https://www.biophysics-reports.org/article/doi/10.52601/bpr.2025.240073]
- 9. Immunoprecipitation Troubleshooting Guide [https://www.cellsignal.com/learn-and-support/troubleshooting/ip-troubleshooting-guide?srsltid=AfmBOorRWdC9odgyZfF4yVmUkMcS2IIYkBqEOrdp9IjOTAugKEL2CjZG]
- 10. Co-immunoprecipitation (Co-IP): The Complete Guide [https://www.antibodies.com/applications/co-immunoprecipitation]
- 11. Advances and challenges in preparing membrane proteins ... [https://www.sciencedirect.com/science/article/abs/pii/S0734975024001770]
- 12. Investigations of Membrane Protein Interactions in Cells Using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11141325/]
- 13. Addressing the challenges in membrane protein ... [https://www.ddw-online.com/analytical-method-to-simplify-the-characterization-of-membrane-proteins-27702-202312/]
- 14. Monitoring endocytosis of integral membrane proteins ... [https://bio-protocol.org/exchange/preprintdetail?id=2855&type=3]
- 15. Immunoprecipitation Troubleshooting Guide [https://www.cellsignal.com/learn-and-support/troubleshooting/ip-troubleshooting-guide?srsltid=AfmBOorpINqFgtMaKcGFVKKTAqyTmpkui5k9SlRs6MdokfZ-pslY_F-w]
- 16. Molecular engineering strategies for visualizing low-affinity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6920673/]
- 17. Large-scale screening for novel low-affinity extracellular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2279249/]
- 18. Protein-Protein Interactions Support—Troubleshooting [https://www.thermofisher.com/sa/en/home/technical-resources/technical-reference-library/protein-assays-analysis-support-center/protein-protein-interactions-support/protein-protein-interactions-support-troubleshooting.html]
- 19. An experimental workflow for enrichment of low abundant ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10062475/]
- 20. Detecting Low Abundance Proteins in Western Blotting [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/detecting-low-abundance-proteins-in-western-blotting.html]
- 21. Western Blot Troubleshooting Guide [https://www.bio-techne.com/resources/protocols-troubleshooting/western-blotting-troubleshooting]
- 22. Immunoprecipitation Troubleshooting Guide [https://www.cellsignal.com/learn-and-support/troubleshooting/ip-troubleshooting-guide?srsltid=AfmBOoo4CdjljQFa3B-yRvP7owWY1J3H6oHCIY5H8AbNw3rJ1zgVBsyF]
- 23. Co-immunoprecipitation (Co-IP) Troubleshooting [https://www.antibodies.com/applications/co-immunoprecipitation/co-ip-troubleshooting]
- 24. Co-immunoprecipitation troubleshooting, tips and tricks [https://www.ptglab.com/news/blog/co-immunoprecipitation-troubleshooting-tips-and-tricks/?srsltid=AfmBOooLN3145TTRc4btCLt4oewBua5L2-nYp_Hz3FrplJn5lLrscQre]
- 25. Common Pull Down Issues and Solutions to Enhance ... [https://www.kmdbioscience.com/article/pull-down-common-problems-and-solutions.html]
- 26. Protein-Protein Interactions Support—Troubleshooting [https://www.thermofisher.com/us/en/home/technical-resources/technical-reference-library/protein-assays-analysis-support-center/protein-protein-interactions-support/protein-protein-interactions-support-troubleshooting.html]
- 27. Comprehensive Identification of Lipid–Membrane Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12044594/]
- 28. Nanodisc single-molecule pulldown to study lipid-protein ... [https://www.sciencedirect.com/science/article/pii/S0022227525001063]
- 29. Evolution of split-protein technologies in virology [https://pmc.ncbi.nlm.nih.gov/articles/PMC12513004/]
- 30. Luciferase complementation for cellular assays beyond ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12064465/]
- 31. NanoBRET: The Bright Future of Proximity-Based Assays [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2019.00056/full]
- 32. BRET (Bioluminescence Resonance Energy Transfer) [https://www.berthold.com/en-us/bioanalytics/knowledge/glossary/bret-bioluminescence-resonance-energy-transfer/]
- 33. Split Luciferase Complementation Assay to Identify Specific ... [https://www.jove.com/v/30485/split-luciferase-complementation-assay-to-identify-specific-protein]
- 34. Protocol for analyzing protein-protein interactions by split ... [https://www.sciencedirect.com/science/article/pii/S2666166724004933]
- 35. Unraveling ER dimerization dynamics in endocrine ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12039421/]
- 36. Protocol to measure and analyze protein interactions in ... [https://www.sciencedirect.com/science/article/pii/S2666166724005136]
- 37. Progress and Prospects in FRET for the Investigation of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12467375/]
- 38. Basics of FRET Microscopy [https://www.microscopyu.com/applications/fret/basics-of-fret-microscopy]
- 39. FRAP, FLIM, and FRET: Detection and analysis of cellular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4515154/]
- 40. FRET‐FLIM to Determine Protein Interactions and Membrane ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11648839/]
- 41. Reliability and accuracy of single-molecule FRET studies ... [https://www.nature.com/articles/s41592-023-01807-0]
- 42. Robust calibration and quantification of FRET signals using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12595091/]
- 43. Membrane extraction with styrene-maleic acid copolymer ... [https://www.nature.com/articles/s41598-022-07606-5]
- 44. Article Functional integrity of membrane protein rhodopsin ... [https://www.sciencedirect.com/science/article/pii/S0006349521004227]
- 45. Optimizing Transmembrane Protein Assemblies in Nanodiscs ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11543783/]
- 46. The styrene–maleic acid copolymer: a versatile tool in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4698303/]
- 47. Molecular Model for the Solubilization of Membranes into ... [https://www.sciencedirect.com/science/article/pii/S0006349514046773]
- 48. Nanodiscs remain indispensable for Cryo-EM studies of ... [https://www.sciencedirect.com/science/article/abs/pii/S0959440X25000600]
- 49. Recent advances in deep learning for protein-protein interaction [https://pmc.ncbi.nlm.nih.gov/articles/PMC12168265/]
- 50. Improved in Silico Identification of Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12021994/]
- 51. A multi-task deep-learning method for predicting membrane ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8650144/]
- 52. ReLo is a simple and rapid colocalization assay to identify ... [https://www.nature.com/articles/s41467-024-47233-4]
- 53. In silico-prediction of protein–protein interactions network ... [https://www.nature.com/articles/s41598-018-33428-5]
- 54. Decoding the limits of deep learning in molecular docking for ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc05395a]
- 55. Quantifying Protein-Protein Interactions of Peripheral ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4119281/]
- 56. Immunoprecipitation Troubleshooting Guide [https://www.cellsignal.com/learn-and-support/troubleshooting/ip-troubleshooting-guide?srsltid=AfmBOorfpTC47Gvpt-gX39faT43Ofim7ws84zRnzwPQclUq23s5PAGTk]
- 57. Protein-Protein Interactions Support—Troubleshooting [https://www.thermofisher.com/my/en/home/technical-resources/technical-reference-library/protein-assays-analysis-support-center/protein-protein-interactions-support/protein-protein-interactions-support-troubleshooting.html]
- 58. Surface Plasmon Resonance: A Versatile Technique for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4481982/]
- 59. Applications of Surface Plasmon Resonance and Biolayer ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9027846/]
- 60. Bio-layer interferometry [https://en.wikipedia.org/wiki/Bio-layer_interferometry]
- 61. Biolayer Interferometry (BLI) [https://cmi.hms.harvard.edu/biolayer-interferometry]
- 62. Surface Plasmon Resonance (SPR) vs Biolayer Interferometry ... [https://www.iaanalysis.com/surface-plasmon-resonance-biolayer-interferometry-choice.html]
- 63. Binding Assays: Common Techniques and Key ... [https://fluidic.com/insight/things-to-consider-when-developing-your-binding-assays/]
- 64. Immunoprecipitation Troubleshooting Guide [https://www.cellsignal.com/learn-and-support/troubleshooting/ip-troubleshooting-guide?srsltid=AfmBOooDgel3ZVz0QaAOlr90Lz2DktGTbnWTEbkJ1NaG6C3Ycozzz4w9]
- 65. Solubilization of proteins: the importance of lysis buffer ... [https://pubmed.ncbi.nlm.nih.gov/26043989/]
- 66. Cell Lysis Methods: A Guide to Efficient Protein Extraction [https://www.bosterbio.com/blog/post/cell-lysis-the-first-and-crucial-step-in-molecular-biology?srsltid=AfmBOooq4zMQHHqirQPegsCS9ulMHax8CNRv7Zyy32VtPNuteOlBiVZr]
- 67. Troubleshooting Guide for NEBExpress E. coli Lysis ... [https://www.neb.com/en-us/tools-and-resources/troubleshooting-guides/troubleshooting-guide-for-nebexpress-e-coli-lysis-reagent-neb-p811?srsltid=AfmBOooNTuLa4FNn3XCOz1F5CrGLNj9N05pjtcJsVKnmJhW9tC-4EaLJ]
- 68. Traditional Methods of Cell Lysis for Protein Extraction [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/traditional-methods-cell-lysis.html]
- 69. Sequential Denaturation and Protein Precipitation assay ... [https://www.sciencedirect.com/science/article/abs/pii/S000326702501116X]
- 70. Comparison of Antibody IgG Binding Proteins [https://www.thermofisher.com/us/en/home/life-science/antibodies/antibodies-learning-center/antibodies-resource-library/antibody-methods/comparison-antibody-igg-binding-proteins.html]
- 71. Choosing Between Proteins A, G, and L: Key Similarities ... [https://www.goldbio.com/blogs/articles/choosing-between-proteins-a-g-and-l-key-similarities-and-differences?srsltid=AfmBOopzWt8biKZ4UVMpfIBdKHZf8OqDHOceFdvZH-dSb_eUO5PdTdvY]
- 72. Affinity of Protein A/G for IgG Types from Different Species [https://www.neb.com/en-us/tools-and-resources/selection-charts/affinity-of-protein-ag-for-igg-types-from-different-species?srsltid=AfmBOoqzpUwj-ePyPPfQ9UQWxVvRaJFbd4FtP6EnmHOsD0tpn4emw8u-]
- 73. ELISA vs Western Blot: When to Use Each Immunoassay ... [https://www.thermofisher.com/blog/life-in-the-lab/western-blot-or-elisa/]
- 74. Western blotting vs. ELISA [https://www.abcam.com/en-us/knowledge-center/western-blot/elisa-vs-western-blot]
- 75. 在细菌表面可分泌表达特殊功能蛋白与免疫球蛋白特异结合 ... [https://www.yeasen.com/h5/solutiondetail/732]
- 76. Selective IgM digestion improves anti-AAV IgG detection in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12416191/]
- 77. Antibody modulation of B cell responses—Incorporating ... [https://www.sciencedirect.com/science/article/pii/S1074761324003157]
- 78. Antibody modulation of B cell responses - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC11257158/]
- 79. Expanding the Reach of Membrane Protein-Ligand ... [https://pubmed.ncbi.nlm.nih.gov/41097828/?utm_source=FeedFetcher&utm_medium=rss&utm_campaign=None&utm_content=1DuyIu5gTabe1jHZSG5UAncFFh_i2RvItg5zrElPBemwI-FEN2&fc=None&ff=20251024114226&v=2.18.0.post22+67771e2]
- 80. A masking clamp for conditional activation of therapeutic ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1640427/full]
- 81. Structure-guided bispecific antibody engineering confers ... [https://www.sciencedirect.com/science/article/pii/S2589004225014361]
- 82. A candidate antibody drug for prevention of malaria [https://www.nature.com/articles/s41591-023-02659-z]
- 83. Isotype Controls [https://www.antibodies.com/primary-antibodies/isotype-controls]
- 84. Isotype Controls - What is it and why use it? Find it out [https://www.lubio.ch/blog/antibody-isotype-controls]
- 85. The emergence of cell-based protein arrays to test for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11346545/]
- 86. Immunoprecipitation and co-immunoprecipitation protocol [https://www.abcam.com/en-us/technical-resources/protocols/immunoprecipitation]
- 87. Immunoprecipitation Troubleshooting Guide [https://www.cellsignal.com/learn-and-support/troubleshooting/ip-troubleshooting-guide?srsltid=AfmBOophAxgUotY_WPHe3MeOvSoAytGa0VsRJE4atcsG4g9Uk-ExCihk]
- 88. Overview of Protease and Phosphatase Inhibition for ... [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/protease-phosphatase-inhibitors.html]
- 89. Challenges in the Development of Functional Assays ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5448992/]
- 90. Phosphatase Inhibitors - an overview [https://www.sciencedirect.com/topics/neuroscience/phosphatase-inhibitors]
- 91. Protease and Phosphatase Inhibitor Tablets [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/protein-biology-application-notes/protease-phosphatase-inhibitor-tablets.html]
- 92. Protease and phoshatase inhibitor cocktails [https://www.abcam.com/en-us/technical-resources/product-overview/protease-and-phosphatase-inhibitor-cocktails]
- 93. Overview of the Immunoprecipitation (IP) Technique [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/immunoprecipitation-ip.html]
- 94. Common Troubleshooting Tips for Western Blot Results [https://www.bosterbio.com/blog/post/common-troubleshooting-tips-for-western-blot-results-part-1?srsltid=AfmBOoqcl46NBVPRaBlIb5kIldtItHA-1qu-zgNkRpStiv2Vng3DKsXr]
- 95. Development and validation of a multiplex bead based ... [https://www.medrxiv.org/content/10.1101/2020.09.02.20185199v1.full-text]
- 96. Validation of a multiplex flow immunoassay for detection of ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0252621]
- 97. Video: Membrane -SPINE: A Biochemical Tool to Identify... [https://www.jove.com/v/50810/membrane-spine-biochemical-tool-to-identify-protein-protein]
- 98. Advances in solubilization and stabilization techniques for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11974516/]
- 99. with Detecting ... | Nature Protocols interactions membrane proteins [https://www.nature.com/articles/nprot.2010.83?error=cookies_not_supported&code=ef579183-7ea9-4924-a192-f299df6a128f]
- 100. - in of vivo - detection on micro-patterned... protein protein interactions [https://pubmed.ncbi.nlm.nih.gov/20305612/]
- 101. PLM-interact: extending protein language models to predict ... [https://www.nature.com/articles/s41467-025-64512-w]
- 102. Review Machine learning to predict de novo protein– ... [https://www.sciencedirect.com/science/article/pii/S0167779925001581]
- 103. Mass spectrometry-based strategies for membrane protein ... [https://www.sciencedirect.com/science/article/abs/pii/S0165614725002366]
- 104. High‐Throughput Pipeline for Protein Expression and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12306927/]
- 105. Non-invasive tools for analysis of plasma membrane ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12103270/]
- 106. Bridging molecular and cellular neuroscience with proximity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12322126/]
- 107. Visualization of lysosomal membrane proteins by cryo ... [https://www.nature.com/articles/s41467-025-64314-0]
- 108. Breaking the boundaries of affinity selection-mass ... [https://www.sciencedirect.com/science/article/pii/S2095177925001960]
- 109. Bacterial two-hybrid systems evolved: innovations for protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12369361/]
- 110. Advanced optical microscopy methods for in situ single ... [https://link.springer.com/article/10.1007/s12551-025-01372-0]
- 111. Membrane proteins structures: A review on computational ... [https://www.sciencedirect.com/science/article/pii/S000527361730233X]
- 112. Challenges In Studying Transient Protein-protein Interactions [https://depixus.com/challenges-in-studying-transient-protein-protein-interactions-for-drug-development-and-how-to-overcome-them/]
- 113. Co-immunoprecipitation: Principles and applications [https://www.abcam.com/en-us/knowledge-center/proteins-and-protein-analysis/co-immunoprecipitation]
- 114. Immunoprecipitation Troubleshooting Guide [https://www.cellsignal.com/learn-and-support/troubleshooting/ip-troubleshooting-guide?srsltid=AfmBOor0TZpfDIOuS84vm4Vq18oaS89idNmHnKkKMAsHH-FNu6T0X5Lt]
- 115. Co-Immunoprecipitation (Co-IP) Technical [https://www.profacgen.com/co-immunoprecipitation-co-ip-technical.htm]
- 116. Troubleshooting CO-IP Assays: Common Problems and ... [https://www.kmdbioscience.com/article/co-ip-assays-common-problems-and-solutions.html]
- 117. Utility of FRET in studies of membrane protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10598290/]
- 118. Protein-Protein Interaction Detection: Methods and Analysis [https://pmc.ncbi.nlm.nih.gov/articles/PMC3947875/]
- 119. Visualization of Protein Interactions in Living Cells - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5788009/]
- 120. SpatialPPIv2: Enhancing protein–protein interaction ... [https://www.sciencedirect.com/science/article/pii/S2001037025000224]
- 121. DPFunc: accurately predicting protein function via deep ... [https://www.nature.com/articles/s41467-024-54816-8]
- 122. Dawn of a New Era for Membrane Protein Design [https://www.sciencedirect.com/science/article/pii/S2693125724000840]
- 123. Predicting and understanding drug-target binding kinetics ... [https://www.cecam.org/workshop-details/predicting-and-understanding-drug-target-binding-kinetics-via-molecular-simulations-1292]
- 124. Mammalian Expression for Membrane Protein Production ... [https://www.thermofisher.com/us/en/home/technical-resources/technical-reference-library/protein-expression-support-center/membrane-protein-support/membrane-protein-support-troubleshooting.html]
- 125. Overcoming research bias: The untapped potential of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12496168/]
- 126. Removing bias against membrane proteins in interaction ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3213014/]
- 127. Challenges in predicting protein-protein interactions of ... [https://www.sciencedirect.com/science/article/pii/S2001037025005057]
- 128. 蛋白的互作验证研究方法简述 [https://www.xiahepublishing.com/2475-7543/MRP-2024-10031]
- 129. 拉曼光谱:膜蛋白相互作用的“live直播镜头” [http://ibp.cas.cn/cb/kpbd/202510/t20251011_7986144.html]
- 130. 創新固定技術突破膜蛋白SPR分析瓶頸助力藥物開發與疾病 ... [https://geneonline.news/%E5%89%B5%E6%96%B0%E5%9B%BA%E5%AE%9A%E6%8A%80%E8%A1%93%E7%AA%81%E7%A0%B4%E8%86%9C%E8%9B%8B%E7%99%BDspr%E5%88%86%E6%9E%90%E7%93%B6%E9%A0%B8%E5%8A%A9%E5%8A%9B%E8%97%A5%E7%89%A9%E9%96%8B%E7%99%BC/]
- 131. Comprehensive Analysis of the Expression, Prognosis and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11986665/]